Why Belly Fat Causes Insulin Resistance (Portal Theory Explained Simply)
Discover how visceral belly fat directly affects your liver through the portal vein, triggering insulin resistance and increasing your risk of type 2 diabetes and fatty liver disease. Learn the science and how to reverse it.
OBESITYDIABETES
Dr. T.S. Didwal, M.D.(Internal Medicine)
4/3/202616 min read
What is the portal theory of insulin resistance?
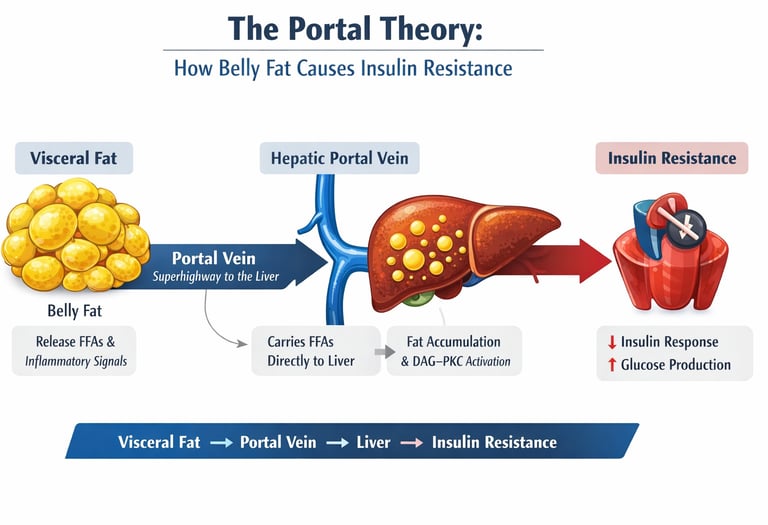
The portal theory posits that visceral (belly) fat releases free fatty acids directly into the hepatic portal vein, delivering them to the liver in high concentrations. This overload triggers fat accumulation in liver cells and disrupts insulin signalling via pathways such as DAG–PKC, leading to hepatic insulin resistance. As a result, the liver continues producing glucose despite normal or high insulin levels, contributing to type 2 diabetes and fatty liver disease
How belly fat causes insulin resistance
Visceral fat releases free fatty acids
Fat enters the portal vein
Liver accumulates fat (DAG)
PKCε blocks insulin signaling
The liver overproduces glucose
Clinical pearls
1. The "First-Pass" Exposure
Scientific Perspective: The liver is the primary site of insulin clearance and glucose regulation. Because visceral fat drains into the portal vein, the liver is exposed to free fatty acids (FFAs) at concentrations 3–4 times higher than the rest of the body.
Think of your liver as a security checkpoint. Usually, it handles a steady stream of traffic. But belly fat is like a "private highway" that dumps a massive traffic jam of fat directly at the liver’s front gate, overwhelming it before the rest of the body can help.
2. The Molecular "Mute" Button (PKCε)
Scientific Perspective: Intracellular accumulation of diacylglycerol (DAG) activates Protein Kinase C epsilon (PKCε). This enzyme phosphorylates the insulin receptor substrate (IRS), effectively decoupling the receptor from its intracellular signaling cascade.
When too much fat enters liver cells, it creates a chemical byproduct that acts like "earplugs" for the cell. Even though your body is screaming at the liver with insulin to stop making sugar, the liver has its earplugs in and can't hear the message.
3. TOFI: The "Hidden" Risk
Scientific Perspective: Thin-Outside-Fat-Inside (TOFI), or Metabolically Obese Normal Weight (MONW), occurs when a patient has low subcutaneous fat but high visceral adiposity. These patients may have a "normal" BMI but present with severe hepatic insulin resistance.
You can’t judge metabolic health by the scale alone. Some people look thin but store all their fat internally around their organs. This "hidden" belly fat is more dangerous than the fat you can actually pinch on your arms or legs.
4. Selective vs. Systemic Resistance
Scientific Perspective: In the early stages, insulin resistance is often pathway-selective. The liver becomes resistant to insulin’s ability to suppress glucose production (gluconeogenesis) but remains sensitive to insulin’s signal to create more fat (lipogenesis), leading to a vicious cycle.
The liver gets confused. It ignores the signal to stop pumping out sugar, but it stays perfectly capable of turning that extra sugar into more liver fat. It’s like a factory that refuses to stop production but keeps building more storage warehouses.
5. Fructose: The Direct-to-Liver Fuel
Scientific Perspective: Unlike glucose, which can be used by every cell in the body, fructose is metabolized almost exclusively in the liver. It bypasses the main rate-limiting step of glycolysis, leading to rapid DNL (de novo lipogenesis) and increased portal FFA load.
While most sugars are used by your muscles for energy, the sugar in sodas and processed sweets (fructose) goes straight to the liver. It’s the fastest way to "fuel" the fat-delivery system that causes insulin resistance.
6. The "Last In, First Out" Rule
Scientific Perspective: Visceral fat is more lipolytically active than subcutaneous fat, meaning it breaks down and enters circulation more easily. However, it is also more metabolically responsive to exercise and caloric deficit.
The good news is that belly fat is "easy come, easy go." While it is the most dangerous fat to have, it is usually the first fat your body burns when you start exercising or improving your diet.
Portal Theory Explained: How Belly Fat Causes Insulin Resistance
Most people think of body fat as a passive energy reserve — something that simply sits under the skin, waiting to be burned. But one specific type of fat behaves very differently. Hidden deep within the abdomen, wrapped around vital organs, visceral fat acts less like storage tissue and more like a metabolically active organ — one that can directly disrupt how your body regulates blood sugar.
What makes visceral fat uniquely dangerous is not just its activity, but its anatomical advantage. Unlike subcutaneous fat, which drains into general circulation, visceral fat empties its contents directly into the liver through the hepatic portal vein. This creates a high-concentration delivery system for free fatty acids (FFAs) and inflammatory signals, exposing the liver to metabolic stress before the rest of the body is affected (Lee & Kim, 2024).
This is the foundation of the portal theory of insulin resistance — a concept that explains why abdominal obesity is so strongly linked to type 2 diabetes and fatty liver disease. According to this model, excess visceral fat increases the flux of FFAs into the liver, overwhelming its metabolic capacity and triggering lipid accumulation within hepatocytes. These lipids, particularly diacylglycerol (DAG), activate intracellular pathways that interfere with insulin signaling, leading to hepatic insulin resistance (Shulman, 2014).
Crucially, this process occurs early — often before fasting glucose levels rise — making it one of the first detectable defects in the progression toward type 2 diabetes. Over time, persistent liver fat accumulation and insulin resistance drive a vicious cycle of increased glucose production, compensatory hyperinsulinemia, and eventual pancreatic dysfunction (Taylor, 2013).
Understanding this liver-centered mechanism changes everything. It shifts the focus from weight alone to fat distribution, from calories to metabolic pathways, and from late-stage disease to early, reversible dysfunction.
What Is Visceral Fat — And Why Does Location Matter?
Body fat is not a passive storage depot. It is an active endocrine organ that secretes hormones, inflammatory signals, and — critically — free fatty acids (FFAs) into the bloodstream.
There are two main types of fat:
Subcutaneous fat: Fat stored just under the skin. This is the fat you can pinch. It drains into the systemic (general body) circulation.
Visceral fat: Fat stored deep inside the abdomen, wrapped around organs like the liver, pancreas, and intestines. This is the fat that causes metabolic trouble.
The critical difference is where visceral fat drains its chemical products. Unlike subcutaneous fat, visceral fat depots — especially the mesenteric fat that surrounds the intestines — drain directly into the hepatic portal vein. This vein is the superhighway that carries blood from the gut and abdominal fat straight to the liver (Lee & Kim, 2024).
This anatomical quirk is the foundation of the portal theory.
The Portal Circulation: Your Gut's Private Highway to the Liver
To understand portal theory, you first need a basic map of the hepatic portal system.
After you eat, nutrients absorbed through your intestinal wall do not enter general circulation right away. Instead, they drain into the portal vein, which carries nutrient-rich blood directly to the liver for processing. The liver acts as the body's first-pass filter — regulating glucose, metabolizing drugs, and managing incoming fatty acids before blood continues to the heart and the rest of the body.
Importantly, the mesenteric (gut-surrounding) fat depots drain into this same portal system. This means that any free fatty acids, hormones, or inflammatory molecules released by visceral fat are delivered at high concentration directly to the liver, before they ever reach the diluting effect of general circulation (Valenzuela-Fuenzalida et al., 2024).
This is very different from subcutaneous fat, whose products are diluted through peripheral circulation before reaching the liver. The liver is therefore uniquely exposed to the metabolic byproducts of visceral fat — and this exposure is the key driver of hepatic insulin resistance.
The Portal Theory of Insulin Resistance: The Core Idea
The portal theory, first articulated and later confirmed with molecular evidence, proposes the following:
Visceral fat releases excess free fatty acids directly into the portal vein → these FFAs flood the liver in high concentrations → the liver becomes insulin resistant → this spreads to the whole body.
This is not just a hypothesis. A landmark study by Kabir et al. (2005) provided direct molecular evidence supporting this causal chain. Using a canine model, the researchers demonstrated that selective removal of visceral (omental) fat significantly improved hepatic insulin sensitivity — while leaving subcutaneous fat intact. The study showed that visceral fat mass was specifically and causally linked to hepatic insulin resistance, not merely associated with it (Kabir et al., 2005).
More recently, Nishizawa et al. (2025) reinforced the significance of mesenteric fat accumulation in glucose dysregulation, noting that mesenteric fat volume is particularly important in determining systemic metabolic outcomes — even beyond what is captured by general measures of obesity like BMI or waist circumference.
Step-by-Step: How Belly Fat Hijacks Your Liver
Here is the portal theory pathway, broken down for clarity:
Step 1: Visceral Fat Releases Free Fatty Acids (FFAs)
When you are overweight — especially with excess abdominal fat — your visceral fat cells (adipocytes) become oversized and metabolically overactive. They release more FFAs into the portal circulation than a healthy liver can manage. This process is called ectopic lipid delivery.
Step 2: FFAs Flood the Liver via the Portal Vein
Because the liver receives portal blood first, it is bombarded with FFAs at concentrations far exceeding what the systemic circulation sees. This is the "portal theory" in action: the physical anatomy of the hepatic portal system gives visceral fat preferential access to the liver.
Step 3: FFAs Activate the DAG–PKC Pathway
This is where the molecular biology becomes critical — and fascinating.
When excess FFAs enter liver cells (hepatocytes), they are converted into diacylglycerol (DAG), a lipid metabolite. DAG accumulation inside the hepatocyte activates an enzyme called protein kinase C epsilon (PKCε).
PKCε is the molecular villain of this story. Once activated, it phosphorylates (interferes with) the insulin receptor substrate proteins (IRS-1 and IRS-2), which are the key signaling molecules that insulin uses to communicate with liver cells. When these substrates are disrupted, the liver cell can no longer "hear" insulin properly (Truong & Lee, 2025; Mancina et al., 2025).
Step 4: Hepatic Insulin Resistance Develops
The result of the DAG–PKCε cascade is hepatic insulin resistance — the liver stops responding normally to insulin. Under normal conditions, insulin signals the liver to suppress glucose production (gluconeogenesis). When the liver is insulin resistant, this suppression fails, and the liver continues producing glucose even after meals and even when blood sugar is already elevated.
This is why people with visceral obesity often have high fasting blood glucose levels — their liver is overproducing glucose despite adequate (or even elevated) insulin levels.
Step 5: The Pancreas Compensates — Until It Can't
The pancreas detects rising blood glucose and secretes more insulin to try to overcome the liver's resistance. This leads to hyperinsulinemia (chronically high insulin levels). Over time, this compensatory effort exhausts the insulin-producing beta cells of the pancreas, setting the stage for type 2 diabetes.
Hepatic Insulin Resistance and Steatotic Liver Disease
The DAG–PKC pathway doesn't just cause metabolic dysfunction — it also drives fat accumulation inside the liver itself.
When the liver cannot properly process the incoming flood of FFAs, it begins storing them as triglycerides within hepatocytes. This is the beginning of metabolic dysfunction-associated steatotic liver disease (MASLD), formerly known as non-alcoholic fatty liver disease (NAFLD).
Truong and Lee (2025) provide detailed mechanistic insight into how hepatic insulin resistance and hepatic steatosis reinforce each other in a vicious cycle: insulin resistance promotes lipogenesis (fat synthesis), and accumulating liver fat worsens insulin resistance further. Their review highlights that this bidirectional relationship makes MASLD both a consequence and an accelerator of metabolic disease (Truong & Lee, 2025).
Genetic factors can also modulate this pathway. Mancina et al. (2025) describe how variants in genes related to lipid metabolism and insulin signaling — including PNPLA3 and TM6SF2 — influence individual susceptibility to steatotic liver disease and insulin resistance, explaining why not everyone with visceral fat develops the same degree of liver dysfunction (Mancina et al., 2025).
From Liver to Heart: The Cardiovascular Connection
The consequences of portal-driven hepatic insulin resistance extend well beyond blood sugar control.
Lee and Kim (2024) provide a comprehensive overview of how visceral adipose tissue dysfunction in cardiometabolic disease operates through multiple overlapping pathways — including inflammation, dyslipidemia (abnormal lipid levels), and endothelial dysfunction — all of which are initiated or amplified by portal FFA delivery to the liver (Lee & Kim, 2024).
A prospective cohort study by Wang et al. (2026) examined the relationship between visceral fat accumulation and cardiovascular disease across different stages of cardiovascular-kidney-metabolic (CKM) syndrome. The researchers found that visceral fat was independently associated with cardiovascular outcomes across all CKM stages, and that arterial stiffness mediated a significant portion of this risk, suggesting that visceral fat's metabolic effects translate into measurable vascular damage (Wang et al., 2026).
This is why central obesity — not just overall body weight — is such a powerful predictor of cardiovascular risk.
Key Takeaways
Visceral fat is not metabolically inert — it functions as an active endocrine organ, releasing free fatty acids (FFAs), cytokines, and adipokines that directly influence systemic metabolism and insulin sensitivity.
The defining feature of visceral fat is its anatomical drainage. Unlike subcutaneous fat, visceral adipose tissue empties into the hepatic portal vein, delivering FFAs to the liver in high concentrations before systemic dilution.
This “first-pass exposure” of the liver is the cornerstone of the portal theory, explaining why central obesity is disproportionately associated with insulin resistance, type 2 diabetes, and metabolic dysfunction-associated steatotic liver disease (MASLD).
Excess portal FFA flux leads to intrahepatic lipid accumulation, particularly diacylglycerol (DAG), which activates protein kinase C epsilon (PKCε), a key inhibitor of insulin receptor signaling.
The result is hepatic insulin resistance, characterized by the liver’s failure to suppress gluconeogenesis despite hyperinsulinemia — a hallmark early defect in metabolic disease progression.
This liver-centered dysfunction precedes systemic abnormalities, often emerging before elevations in fasting glucose or HbA1c, making it a critical but under-recognized therapeutic target.
A self-reinforcing metabolic cycle develops: hepatic fat accumulation worsens insulin resistance, which in turn promotes further lipogenesis and glucose overproduction.
Beyond glycemic control, portal-driven hepatic dysfunction contributes to dyslipidemia, increased VLDL production, and downstream cardiovascular risk through endothelial dysfunction and arterial stiffness.
Importantly, this process is highly modifiable. Reduction in visceral fat through targeted lifestyle interventions can rapidly decrease hepatic fat content and restore insulin sensitivity.
Clinical focus must shift from total adiposity to fat distribution, emphasizing waist circumference, liver health, and metabolic markers over BMI alone.
The portal theory reframes metabolic disease as a liver-first pathology, offering a mechanistic bridge between obesity, fatty liver, and type 2 diabetes.
Understanding this pathway enables earlier, more precise intervention, transforming both prevention strategies and long-term cardiometabolic outcomes.
Controversy & Nuance While the portal theory highlights direct free fatty acid delivery to the liver as a key driver of insulin resistance, it is not the sole mechanism. Systemic inflammation, adipokine imbalance (e.g., low adiponectin), and ectopic fat deposition in muscle and liver also play important, interacting roles in metabolic dysfunction.
Practical Applications: What You Can Do Right Now
Understanding the portal theory is not just an academic exercise. It offers concrete, actionable guidance for reducing visceral fat and breaking the cycle of hepatic insulin resistance.
1. Target Visceral Fat Specifically
Not all weight loss is metabolically equal. Aerobic exercise — particularly moderate-intensity sustained cardio — has been shown to preferentially reduce visceral fat compared to subcutaneous fat. Even a modest 5–10% reduction in body weight can significantly reduce portal FFA delivery to the liver.
2. Reduce Refined Carbohydrates and Added Sugars
Fructose, found abundantly in sugary drinks and processed foods, is metabolized almost entirely in the liver and directly promotes hepatic lipogenesis (fat production), worsening hepatic insulin resistance independent of total caloric intake. Replacing refined carbohydrates with complex carbohydrates and fiber is one of the most evidence-based dietary strategies for improving hepatic insulin sensitivity.
3. Prioritize Resistance Training
Building muscle mass improves whole-body insulin sensitivity and acts as a glucose sink, reducing the metabolic burden on the liver. Resistance training 2–3 times per week is recommended alongside aerobic activity.
4. Improve Sleep Quality
Sleep deprivation and circadian disruption increase cortisol and growth hormone dysregulation, which promote visceral fat accumulation and exacerbate insulin resistance. Aiming for 7–9 hours of quality sleep per night is a legitimate metabolic intervention.
5. Reduce Visceral Stress (Literally)
Chronic psychological stress elevates cortisol, which specifically drives visceral fat deposition. Stress management strategies — including mindfulness, structured relaxation, and social connection — may have direct metabolic benefits.
6. Monitor the Right Numbers
Standard weight and BMI measurements do not capture visceral fat distribution. Ask your doctor about:
Waist circumference (goal: <80 cm for women, <94 cm for men in most guidelines)
Fasting insulin and HOMA-IR (insulin resistance index)
Liver enzymes (ALT, AST) as markers of hepatic stress
Triglyceride: HDL ratio as a clinical surrogate for insulin resistance
7. Consider Time-Restricted Eating
Emerging evidence suggests that aligning eating windows with circadian biology (e.g., eating within an 8–10 hour daytime window) may improve hepatic insulin sensitivity and reduce visceral fat, independent of caloric restriction.
Frequently Asked Questions
Q1. What exactly is the portal theory of insulin resistance? The portal theory proposes that visceral (belly) fat releases free fatty acids directly into the hepatic portal vein, delivering them in high concentrations to the liver. This overwhelms the liver's fat-processing capacity, triggers the DAG–PKCε pathway, and causes the liver to stop responding properly to insulin — a condition called hepatic insulin resistance. This then spreads systemically, contributing to type 2 diabetes and cardiovascular disease.
Q2. Why is visceral fat more dangerous than fat elsewhere on the body? It comes down to anatomy. Visceral fat drains directly into the portal vein that feeds the liver, bypassing the diluting effect of general circulation. Subcutaneous (under-skin) fat, by contrast, drains into general circulation and does not expose the liver to the same high-concentration FFA load. The liver's unique vulnerability to portal-delivered fat makes visceral fat disproportionately harmful metabolically.
Q3. What is the DAG–PKC pathway and why does it matter? DAG (diacylglycerol) is a fat metabolite that accumulates inside liver cells when too many fatty acids arrive from portal blood. DAG activates an enzyme called PKCε (protein kinase C epsilon), which then blocks the insulin signaling cascade inside the hepatocyte. The result: the liver cannot hear insulin's message to stop producing glucose. This is the molecular mechanism by which belly fat causes blood sugar to rise.
Q4. Can you have a normal BMI and still have dangerous visceral fat? Yes — this is a phenomenon called "metabolically obese normal weight" (MONW) or "thin-fat" syndrome. People with a normal BMI but high waist-to-height ratio, or high visceral fat volume detected on imaging, can have the same hepatic insulin resistance as someone with a higher BMI. This is why measuring waist circumference and metabolic markers (fasting insulin, triglycerides, liver enzymes) is more informative than weight alone.
Q5. Does the portal theory explain fatty liver disease, too? Yes, directly. When the liver is overwhelmed by portal FFA delivery, it cannot fully oxidize (burn) all incoming fats, so it begins storing them as triglycerides within liver cells. This is the beginning of metabolic dysfunction-associated steatotic liver disease (MASLD). Insulin resistance worsens hepatic fat accumulation, and hepatic fat worsens insulin resistance — creating a self-reinforcing cycle (Truong & Lee, 2025).
Q6. Is hepatic insulin resistance reversible? Encouragingly, yes — it is highly responsive to lifestyle intervention. Studies consistently show that aerobic exercise, dietary improvement (particularly reducing refined carbohydrates and fructose), and even modest weight loss (5–10%) can significantly reduce hepatic fat content and restore insulin sensitivity within weeks to months. Early intervention is key, before fibrosis (scarring) develops in the liver.
Q7. What blood tests should I ask for to check if I have hepatic insulin resistance? There is no single perfect test, but a useful panel includes: fasting glucose and insulin (to calculate HOMA-IR), liver enzymes (ALT, AST — elevated in fatty liver), lipid panel with attention to triglycerides and HDL-cholesterol, and a triglyceride:HDL ratio above 3.0 is considered a strong clinical indicator of insulin resistance. If your doctor suspects significant liver fat, a liver ultrasound or FibroScan can be performed non-invasively.
Q8. How does visceral fat cause insulin resistance?
Visceral fat releases excess fatty acids into the portal circulation. These accumulate in the liver as diacylglycerol (DAG), activating PKCε, which blocks insulin signaling and leads to hepatic insulin resistance.
Q9. Can you be thin and still have insulin resistance?
Yes. This is called “metabolically obese normal weight.” Even with a normal BMI, high visceral fat can impair liver insulin sensitivity and increase diabetes risk.
Q10. What is the fastest way to reduce visceral fat?
The most effective strategy combines:
Aerobic exercise (especially Zone 2)
Resistance training
Reduced refined carbohydrates and sugar
Even a 5–10% weight loss significantly reduces liver fat.
Call to Action: Take the Next Step
Understanding the portal theory is empowering — because it tells you exactly where the problem is and exactly what to target.
Your 3-Step Action Plan This Week:
Step 1 — Know Your Numbers. Book an appointment with your doctor or healthcare provider and ask for: fasting glucose, fasting insulin (HOMA-IR), ALT/AST, and a full lipid panel. If you have a tape measure at home, check your waist circumference today.
Step 2 — Start Moving Your Liver. Commit to 30 minutes of moderate aerobic activity — brisk walking, cycling, or swimming — at least 5 days this week. This is the single most effective non-pharmacological intervention for reducing visceral fat.
Step 3 — Upgrade One Meal. Replace your most sugar-heavy meal or snack of the day with something high in protein, fiber, and healthy fat. This small change directly reduces the fructose and refined carbohydrate load hitting your liver through the portal vein.
Author’s Note
As clinicians, we are often trained to interpret metabolic disease through numbers — glucose levels, HbA1c, lipid panels. But behind these values lies a deeper, more dynamic physiology that is not always immediately visible in routine practice. One of the most important shifts in modern metabolic medicine has been the recognition that where fat is stored matters as much as how much fat is present.
This article was written to bridge that gap — to translate a complex, research-driven concept like the portal theory of insulin resistance into a framework that is both scientifically rigorous and clinically meaningful. The goal is not only to explain what happens, but to clarify why it happens and, most importantly, what can be done about it.
The evidence is increasingly clear: hepatic insulin resistance is an early, central defect in the development of type 2 diabetes and cardiometabolic disease. Yet, it is also highly modifiable, particularly in its early stages. Interventions that reduce visceral fat, improve hepatic lipid handling, and restore insulin signaling can fundamentally alter disease trajectory — often more powerfully than patients realize.
For healthcare professionals, this underscores the importance of moving beyond BMI-centric models toward a more nuanced assessment of metabolic risk. For patients, it offers something equally valuable: a sense of control. The mechanisms described here are not abstract biochemical pathways — they are targets for real-world intervention.
If this article helps shift even one conversation from “weight loss” to metabolic health, or prompts earlier detection and intervention, it has served its purpose.
Share This Article If It Helped You
If this explanation helped you understand why belly fat is dangerous, share it with someone who's been told they have "pre-diabetes," "fatty liver," or "metabolic syndrome." This is exactly the kind of mechanistic understanding that can change how people approach their health.
Join the Conversation
Have a question about your metabolic health? Drop it in the comments below. I read and respond to every question personally.
Disclaimer: This article is for informational purposes only and does not constitute medical advice. Individual circumstances vary, and treatment decisions should always be made in consultation with qualified healthcare professionals.
Related Articles
The #1 Diet Strategy to Reduce Visceral Fat According to Latest Research
How to Lose Visceral Fat Fast: The Science of HIIT, Zone 2, and Strength Training
Is Your Muscle Insulin Resistant? 2026 Update | DR T S DIDWAL
Activate Your Brown Fat: A New Pathway to Longevity and Metabolic Health | DR T S DIDWAL
https://drdidwal.com/beyond-bmi-why-waist-to-height-ratio-whtr-is-the-new-clinical-gold-standard
What Causes Visceral Fat? Hormones, Lifestyle, and Metabolic Risk Explained | DR T S DIDWAL
The BMI Paradox: Why "Normal Weight" People Still Get High Blood Pressure | DR T S DIDWAL
References
Kabir, M., Catalano, K. J., Ananthnarayan, S., Kim, S. P., Van Citters, G. W., Dea, M. K., & Bergman, R. N. (2005). Molecular evidence supporting the portal theory: A causative link between visceral adiposity and hepatic insulin resistance. American Journal of Physiology-Endocrinology and Metabolism, 288(2), E454–E461. https://doi.org/10.1152/ajpendo.00203.2004
Lee, M.-J., & Kim, J. (2024). The pathophysiology of visceral adipose tissues in cardiometabolic diseases. Biochemical Pharmacology, 222(Suppl C), Article 116116. https://doi.org/10.1016/j.bcp.2024.116116
Mancina, R. M., Valenti, L., & Romeo, S. (2025). Human genetics of steatotic liver disease: Insights into insulin resistance and lipid metabolism. Nature Metabolism, 7, 2199–2211. https://doi.org/10.1038/s42255-025-01394-8
Nishizawa, H., Nagao, H., & Shimomura, I. (2025). Visceral fat accumulation and glucose metabolism: Significance of mesenteric fat mass. The Journal of Clinical Endocrinology & Metabolism, 110(12), e4234–e4235. https://doi.org/10.1210/clinem/dgaf012
Truong, X. T., & Lee, D. H. (2025). Hepatic insulin resistance and steatosis in metabolic dysfunction-associated steatotic liver disease: New insights into mechanisms and clinical implications. Diabetes & Metabolism Journal, 49(5), 964–986. https://doi.org/10.4093/dmj.2025.0644
Valenzuela-Fuenzalida, J. J., Rodríguez-Osorio, B., Salgado-Torres, C., et al. (2024). A systematic review and meta-analysis: Prevalence and clinical implications of anatomical variants of the hepatic portal vein. Scientific Reports, 14, Article 30002. https://doi.org/10.1038/s41598-024-81543-3
Wang, K., Luo, X., Wan, D., et al. (2026). Associations of visceral fat accumulation with cardiovascular disease across cardiovascular-kidney-metabolic syndrome stages 0–3: Mediation effects of arterial stiffness in a prospective cohort study. BMC Cardiovascular Disorders. https://doi.org/10.1186/s12872-026-05775-z
Taylor, R. (2013). Type 2 diabetes: Etiology and reversibility. Diabetes Care, 36(4), 1047–1055. https://doi.org/10.2337/dc12-1805
Shulman, G. I. (2014).Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. New England Journal of Medicine, 371(12), 1131–1141.
https://doi.org/10.1056/NEJMra1011035