Muscle Insulin Resistance: The Hidden Signaling Failure Behind Metabolic Disease
Understand how skeletal muscle insulin resistance develops at the molecular level—IRS-1 disruption, PI3K-Akt failure, inflammation, and lipotoxicity.
METABOLISM
Dr. T.S. Didwal, M.D.(Internal Medicine)
3/8/202616 min read
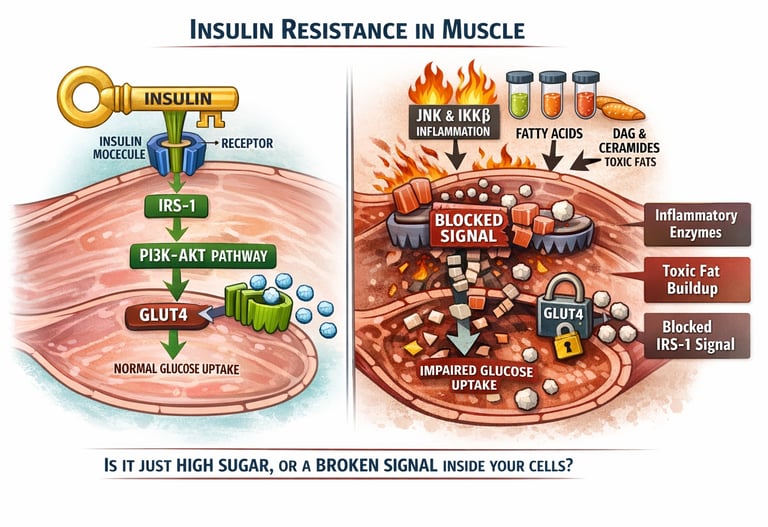
Why Muscle Insulin Resistance Appears Before Diabetes
Skeletal muscle is responsible for nearly 70–80% of post-meal glucose disposal. When muscle cells become resistant to insulin, glucose uptake falls early, forcing the pancreas to produce more insulin. Over time, this compensatory hyperinsulinemia progresses toward prediabetes and type 2 diabetes
When people hear the words insulin resistance, they usually think of high blood sugar. But the real problem begins much earlier — deep inside your muscle cells, long before glucose levels start to rise.
Every time you eat, insulin acts like a messenger. Its job is simple: tell your muscle cells to open their doors and absorb glucose from your bloodstream. In fact, skeletal muscle is responsible for clearing nearly 70–80% of glucose after a meal. When this system works well, blood sugar remains stable and your cells are efficiently fueled.
But in insulin resistance, the message gets distorted.
At the molecular level, insulin’s signal travels through a carefully organized chain of proteins. One of the first and most important is a molecule called insulin receptor substrate-1 (IRS-1). When IRS-1 functions properly, it activates a pathway known as PI3K–Akt — the main route that allows glucose transporters (GLUT4) to move to the cell surface and pull sugar inside.
However, research now shows that subtle chemical modifications to IRS-1 — particularly a process called serine phosphorylation — can block this signal at its earliest step (Woo et al., 2024). Inflammatory enzymes such as JNK and IKKβ, often activated by excess fatty acids and metabolic stress, further suppress this pathway (Dutta, 2025). At the same time, fat-derived molecules like diacylglycerol and ceramides accumulate inside muscle cells and interfere directly with insulin signaling (Tang et al., 2026).
The result? Insulin is present. The receptors are present. But the internal machinery that carries the message forward begins to fail.
Understanding what your muscle cells are actually doing — and why — is the first step toward reversing insulin resistance before it progresses to type 2 diabetes.
Clinical pearls
1. The "Handshake" Sabotage (IRS-1 Phosphorylation)
Scientific Perspective: Metabolic stress activates kinases like JNK and IKKβ, which catalyze the phosphorylation of IRS-1 at serine residues rather than tyrosine. This structural modification creates steric hindrance, preventing IRS-1 from docking with the insulin receptor, effectively "uncoupling" the receptor from its downstream metabolic effects.
Think of insulin and its receptor as two people trying to shake hands to start a job. When you are under metabolic stress, your body puts a 'glove' of inflammatory signals over the hand. The two can’t feel each other or grip, so the handshake never happens, and the work of clearing sugar never starts."
2. The BCAA "Traffic Jam" (The High-Protein Paradox)
Scientific Perspective: As highlighted in Li et al. (2026), an oversupply of branched-chain amino acids (BCAAs), particularly in a sedentary context, can overactivate the mTOR/S6K1 pathway. This leads to feedback inhibition of IRS-1 via serine phosphorylation, proving that even "healthy" macronutrients can drive insulin resistance if they exceed the muscle's oxidative demand.
"While protein is vital for muscle, your body can have too much of a good thing. If you eat a very high-protein diet but don't exercise enough to burn those building blocks, they start to 'pile up' at the factory gates. This pile-up actually signals the cell to stop taking in sugar, paradoxically making you more insulin resistant."
3. Lipotoxicity: The "Oil in the Gears" (DAGs & Ceramides)
Scientific Perspective: It is not total intramyocellular triacylglycerol (IMTG) that impairs signaling, but the accumulation of bioactive intermediates: Diacylglycerol (DAG) and Ceramides. DAGs activate PKCθ, which inhibits IRS-1, while Ceramides activate PP2A, which directly dephosphorylates and inactivates Akt, the master regulator of glucose transport.
"Not all fat in the muscle is bad, but certain 'toxic' fats act like sand in the gears of a watch. These molecules (DAGs and Ceramides) specifically break the internal machinery that moves sugar transporters to the surface. It’s not just about 'being fat'; it’s about the specific type of fat 'exhaust' clogging your cells."
4. The "Inflammatory Fire" (JNK & IKKβ)
Scientific Perspective: Chronic low-grade inflammation acts through the JNK and IKKβ pathways to create a self-perpetuating loop. These kinases don't just block insulin signaling; they activate NF-κB, which travels to the nucleus to produce more inflammatory cytokines (TNF-α, IL-6), ensuring the "jammed lock" stays jammed even if blood sugar temporarily drops.
"Insulin resistance isn't just a sugar problem; it's an 'internal fire' problem. These inflammatory signals act like a persistent alarm bell ringing inside your cells. As long as that alarm is going off, your cells stay in 'defense mode' and refuse to open the doors for sugar, even if the pancreas is screaming at them with more insulin."
5. The "Bypass Road" (AMPK vs. Akt)
Scientific Perspective: The PI3K-Akt highway is the primary route for insulin-stimulated glucose uptake and is the main site of failure in diabetes. However, the AMPK pathway (activated by muscle contraction and energy depletion) remains a functional "bypass," capable of mobilizing GLUT4 to the membrane even when the Akt highway is completely obstructed by ceramides or inflammatory kinases.
"When the main highway (the insulin pathway) is closed due to a wreck, your muscles have a secret 'back road.' This road only opens when you physically move. Even if your insulin signaling is badly damaged, taking a walk or lifting weights uses this alternative route to get the sugar out of your blood and into your muscles."
Part 1: What Is IRS-1 and Why Does It Matter?
Insulin Receptor Substrate 1, or IRS-1, is one of the first proteins to receive the insulin signal after insulin binds to its receptor on a muscle cell. Think of IRS-1 as a relay runner in a biochemical relay race. When insulin arrives, the insulin receptor "hands the baton" to IRS-1, which then carries the signal forward to activate glucose transport and other metabolic processes.
For IRS-1 to work properly, it needs to be activated by a process called tyrosine phosphorylation — the addition of a phosphate group to specific tyrosine (an amino acid) sites on the protein. This is the normal, healthy activation signal. However, when the wrong sites on IRS-1 get phosphorylated — specifically the serine residues — the protein is essentially turned off, and the insulin signal is blocked.
The 2024 Breakthrough: Structural Evidence of Signaling Failure
1. The "Handshake" Disruption (Woo et al., 2024)
The Discovery: Using high-resolution mass spectrometry (HDX-MS), researchers mapped the exact structural change that causes insulin resistance.
The Mechanism: Chemical modification (phosphorylation) of specific serine sites within the IRS-1 protein physically changes its shape.
The Result: IRS-1 can no longer dock or "handshake" with the insulin receptor. Because the physical connection is blocked, the insulin signal cannot be passed into the cell. The relay baton is dropped at the very first step.
2. Why the Relay Fails (Pathological Triggers)
Serine phosphorylation is the body's molecular "stress response" to several environmental and biological factors:
Lipid Excess: High levels of circulating free fatty acids.
Systemic Inflammation: Elevated cytokines like TNF-α and IL-6 (often seen in obesity and sedentary lifestyles).
Cellular Stress: Overactivity of "stress kinases" like JNK and IKKβ, which act as the enzymes that physically attach the blocking phosphate groups to IRS-1.
3. The Downstream Consequence
Without the IRS-1 "handshake," the entire internal message system is severed. This is why, even when insulin levels are high, the muscle cell remains "deaf" to the signal, leading to:
Inability to absorb glucose.
Persistent high blood sugar.
Compensatory hyperinsulinemia (the body overproducing insulin to try to force the message through).
Part 2: PI3K–Akt Pathway Disruption — The Highway That Gets Blocked
The Normal Pathway: Insulin Signaling at Its Best
Once IRS-1 is properly activated by the insulin receptor, it recruits a critical enzyme called phosphatidylinositol 3-kinase (PI3K). PI3K then produces a lipid messenger (PIP3) that activates a protein kinase called Akt (also known as protein kinase B). Akt, when activated, sets off a powerful chain of metabolic events:
It moves glucose transporter 4 (GLUT4) vesicles to the cell surface, allowing glucose to enter the muscle cell
It stimulates glycogen synthesis (storage of glucose as glycogen)
It promotes protein synthesis and inhibits protein breakdown
In a healthy metabolic state, this PI3K–Akt highway is the main route by which insulin delivers glucose to your muscles. When it is working efficiently, blood glucose is rapidly controlled after every meal.
How the Pathway Gets Derailed
This breakdown, based on the Tang et al. (2026) review, illustrates the systematic failure of the insulin signaling "relay race." When these four points are disrupted, the result is functional insulin resistance in the muscle cell.
The 4 Stages of Insulin Signaling Failure
Depleted "Relay Runners" (IRS-1 Suppression)
The Science: Chronic inflammation triggers signaling pathways that suppress the production of the IRS-1 protein.
The Result: With fewer IRS-1 proteins available, the cell cannot effectively catch the signal sent by the insulin receptor.
Recruitment Failure (Decreased PI3K Activity)
The Science: Without a strong IRS-1 signal, the enzyme PI3K is not recruited to the cell membrane.
The Result: This leads to a drop in PIP3 production, which is the essential chemical "spark" needed to move the signal deeper into the cell.
The "Off Switch" Overload (Impaired Akt Phosphorylation)
The Science: Even if some signal reaches Akt, opposing proteins (phosphatases) act like an "off switch" by stripping away the phosphate groups needed to activate it.
The Result: Akt remains dormant or underactive, stalling the command to move glucose.
The Locked Gate (GLUT4 Translocation Failure)
The Science: This is the functional endpoint. Reduced Akt activity means the GLUT4 glucose transporters remain trapped inside the cell rather than moving to the surface.
The Result: The "gates" stay locked, and glucose remains stuck in the bloodstream instead of being absorbed by the muscle.
Why This Matters for Patients
The PI3K–Akt pathway is not just an academic concept — its disruption has direct, measurable consequences:
Higher fasting and post-meal blood glucose levels
Reduced muscle glycogen stores, affecting energy and exercise performance
Compensatory hyperinsulinemia (the pancreas releases more insulin to try to overcome the resistance)
Progressive pancreatic beta-cell exhaustion over time, eventually leading to type 2 diabetes
Understanding this pathway is also clinically useful because it reveals why certain lifestyle interventions — particularly exercise — are so powerfully therapeutic. Muscle contractions can activate GLUT4 translocation and Akt through independent, insulin-bypassing mechanisms (such as AMPK activation), essentially creating a "back road" around the blocked highway.
Part 3: Inflammatory Kinases (JNK and IKKβ) — The Fire Inside Your Muscle Cells
Inflammation as a Molecular Saboteur
Chronic low-grade inflammation — the kind that does not cause obvious symptoms but quietly damages tissues — plays a central role in developing insulin resistance. Two inflammatory kinases, JNK and IKKβ, are among the most powerful molecular saboteurs involved.
These kinases are activated by a wide range of metabolic stressors: saturated fats, inflammatory cytokines, reactive oxygen species (oxidative stress), and excess branched-chain amino acids (BCAAs). Once activated, both JNK and IKKβ directly phosphorylate IRS-1 at serine residues — particularly serine-307 in rodents (a critical regulatory site), shutting down insulin signaling at its very first step.
The JNK/IKKβ Complex: The Molecular Brakes on Insulin
1. The JNK "Feed-Forward" Cycle
The Mechanism: JNK (c-Jun N-terminal kinase) is a stress-activated protein that links lipid excess and inflammation to metabolic failure.
The Damage: JNK performs "inhibitory phosphorylation" on IRS-1, effectively cutting the insulin signaling cable.
The 2025 Insight: Research highlights that JNK in skeletal muscle is self-amplifying. Once activated by obesity or inflammation, it creates a loop that keeps insulin signaling suppressed even after the initial trigger is removed.
2. The High-Protein Paradox (2026 Finding)
The Science: While high-protein diets are often used for weight loss, excess protein can elevate BCAAs (Branched-Chain Amino Acids) in the blood.
The Impact: These elevated amino acids can paradoxically activate the JNK/IKKβ pathway.
The Takeaway: In the wrong metabolic context, excess protein can worsen insulin resistance by triggering the same "molecular brakes" as high-fat diets.
3. IKKβ and the Self-Sustaining Inflammatory Loop
Dual Damage: IKKβ damages insulin sensitivity in two ways:
Directly: It shuts down IRS-1 (like JNK).
Indirectly: It activates NF-κB, the "master switch" of inflammation.
The Result: NF-κB produces inflammatory cytokines (TNF-α, IL-6) that circle back to activate more JNK and IKKβ.
4. Why Persistence Matters
Because this inflammatory machinery is self-sustaining, reversing insulin resistance isn't just about cutting calories. It requires anti-inflammatory interventions—such as exercise, restorative sleep, and stress management—to manually "reset" these overactive molecular switches.
Part 4: Lipotoxic Intermediates (DAG and Ceramides) — When Fat Becomes Toxic to Muscle
This brief note summarizes the Lipotoxicity Concept, explaining how specific fat metabolites (DAGs and Ceramides) physically sabotage the insulin signaling machinery within muscle cells.
The Lipotoxicity Concept: When Fat Becomes Toxic
Lipotoxicity is the process where excess fat-derived molecules accumulate inside muscle cells and exert "toxic" effects on metabolism. It is not the amount of fat that matters most, but the accumulation of bioactive lipid intermediates.
1. Diacylglycerol (DAG): The PKC Activator
The Mechanism: When the delivery of fat to muscle cells exceeds the cell's ability to burn it (oxidation), DAG levels rise.
The Sabotage: Elevated DAG activates Protein Kinase C (PKC) isoforms (specifically PKCθ and PKCε).
The Result: Like JNK and IKKβ, these PKCs perform inhibitory serine phosphorylation on IRS-1, "cutting the wire" of the insulin signal.
The Exercise Fix: Regular exercise increases the muscle's ability to burn fat, clearing DAG out of the cell and restoring insulin sensitivity.
2. Ceramides: The Potent Multi-Threat
The Mechanism: Ceramides are synthesized inside muscle cells when there is an excess of saturated fatty acids (like palmitate).
The Sabotage: Ceramides are more potent than DAG because they attack the insulin pathway from multiple angles:
Direct Inhibition: They prevent the activation of Akt, the key enzyme that tells the cell to absorb glucose.
Mitochondrial Stress: They disrupt energy production, making the cell even less efficient at burning fat.
The Dietary Link: Diets high in processed meats and fried foods directly fuel ceramide production, explaining why saturated fat can worsen insulin resistance even before weight gain occurs.
3. The "Metabolic Mismatch"
Total Fat vs. Bioactive Fat: This explains why two people with the same weight can have different metabolic health. The person who burns their fat stores through activity prevents these "toxic" intermediates from building up.
The End Result: Whether via DAG or Ceramides, the muscle cell becomes "clogged," the GLUT4 gates remain locked, and blood sugar rises.
Mitochondrial Dysfunction in Muscle Insulin Resistance (Brief Note)
Mitochondrial dysfunction is increasingly recognized as a central contributor to skeletal muscle insulin resistance. When mitochondrial oxidative capacity declines, the muscle cell becomes less efficient at β-oxidation, the process that breaks down fatty acids to generate energy. As a result, fatty acids entering the muscle are not fully metabolized, leading to incomplete fatty-acid oxidation and accumulation of lipid intermediates.
This metabolic bottleneck promotes the buildup of bioactive lipids such as diacylglycerol (DAG) and ceramides, which directly interfere with insulin signaling pathways. DAG activates protein kinase C (PKC) isoforms that inhibit IRS-1 function, while ceramides suppress Akt activation, blocking GLUT4-mediated glucose uptake.
At the same time, dysfunctional mitochondria produce higher levels of reactive oxygen species (ROS). Excess ROS amplifies cellular stress and activates inflammatory kinases such as JNK and IKKβ, further impairing insulin signaling.
Thus, impaired mitochondrial fat oxidation creates a convergence of lipotoxicity, oxidative stress, and inflammatory signaling, all of which reinforce skeletal muscle insulin resistance. Improving mitochondrial function through regular exercise and metabolic conditioning can enhance fatty-acid oxidation, reduce DAG and ceramide accumulation, and restore insulin sensitivity.
Current and Emerging Therapeutic Implications
Understanding these molecular mechanisms is not merely academic — it has direct implications for how we treat and prevent insulin resistance.
Exercise remains the most powerful known intervention. Muscle contractions independently activate GLUT4 translocation via AMPK, bypass disrupted IRS-1/PI3K/Akt signaling, reduce intramyocellular DAG and ceramide content by enhancing fat oxidation, and suppress chronic inflammatory kinase activity with consistent training.
Dietary composition matters profoundly at the molecular level. Reducing saturated fat intake directly reduces ceramide and DAG synthesis. The evidence on high-protein diets, highlighted by Li et al. (2026), urges caution in recommending very high-protein diets to insulin-resistant patients without individualizing the approach.
Anti-inflammatory pharmacotherapy targeting JNK and IKKβ is an active area of drug development, though clinical translation has been challenging due to the broad physiological roles of these kinases.
Sphingolipid metabolism targeting — specifically pharmacological reduction of ceramide synthesis — represents a promising frontier in metabolic drug development.
Key Takeaways: Molecular Insulin Resistance in Skeletal Muscle
Skeletal muscle is the primary glucose sink. Nearly 70–80% of insulin-stimulated glucose disposal occurs in skeletal muscle. When muscle becomes insulin resistant, whole-body glucose control destabilizes early — often years before type 2 diabetes is diagnosed.
The problem begins at the signaling level — not the sugar level. Insulin resistance is fundamentally a signaling defect. Insulin may be present in normal or even high amounts, but the intracellular communication cascade fails.
IRS-1 dysfunction is an early molecular lesion. In healthy cells, insulin activates IRS-1 through tyrosine phosphorylation. In insulin resistance, inhibitory serine phosphorylation disrupts IRS-1’s ability to transmit the signal forward, effectively “dropping the baton” at the start of the pathway.
The PI3K–Akt pathway is the metabolic highway. When IRS-1 signaling falters, PI3K activation declines, Akt phosphorylation weakens, and GLUT4 transporters fail to translocate to the membrane — directly impairing glucose uptake.
Inflammatory kinases are central drivers. Stress-responsive enzymes such as JNK and IKKβ, activated by excess fatty acids, cytokines, and oxidative stress, phosphorylate IRS-1 at inhibitory sites and sustain a feed-forward inflammatory loop.
Lipotoxic intermediates amplify the damage. Intramyocellular accumulation of diacylglycerol (DAG) activates PKC isoforms that impair IRS-1, while ceramides inhibit Akt through PP2A activation — attacking the pathway at multiple nodes simultaneously.
Mechanisms are interconnected, not isolated. Lipid accumulation, inflammatory signaling, mitochondrial stress, and impaired glucose transport reinforce one another, creating a self-perpetuating metabolic network.
Reversibility is biologically plausible. Exercise enhances GLUT4 translocation independent of insulin, increases fatty acid oxidation, reduces ceramide/DAG burden, and suppresses inflammatory kinase activity — directly targeting core molecular defects.
Diet quality matters at the molecular level. Saturated fat excess promotes ceramide synthesis; extreme macronutrient patterns may influence JNK/IKKβ signaling. Composition is as important as calories.
Clinical implication: Insulin resistance is not a single defect but a systems-level signaling disruption — and effective intervention must address inflammation, lipid handling, and cellular signaling simultaneously.
Frequently Asked Questions (FAQs)
Q1: What is the simplest way to understand insulin resistance in muscle?
Think of it this way: insulin is supposed to work like a key that unlocks muscle cells so sugar can enter and provide energy. In insulin resistance, the "lock" inside the cell is jammed — not by one problem but by several: inflammatory signals, toxic fat molecules, and disrupted protein activity. The key (insulin) may still be present, but the door stays closed. Blood sugar then builds up in the bloodstream instead of being used for energy.
Q2: Is insulin resistance in muscle reversible?
Yes, to a meaningful degree — particularly in the earlier stages. Lifestyle interventions, especially regular aerobic and resistance exercise, have been shown to reduce intramyocellular DAG and ceramide levels, improve PI3K–Akt signaling, and suppress inflammatory kinase activity. Dietary quality improvements — particularly reducing saturated fats and ultra-processed foods — also directly address several of the molecular mechanisms described in this article.
Q3: Why is skeletal muscle specifically so important in insulin resistance?
Because skeletal muscle accounts for approximately 70–80% of insulin-stimulated glucose uptake in the body. When this tissue becomes resistant, the burden on the pancreas to produce more insulin increases dramatically, and blood glucose regulation deteriorates. No other tissue has as large an impact on whole-body glucose metabolism.
Q4: Can a high-protein diet cause insulin resistance?
Emerging evidence, particularly from Li et al. (2026), suggests that chronically excessive protein intake may activate the JNK/IKKβ–IRS-1 pathway and worsen insulin sensitivity — particularly when the protein is rich in branched-chain amino acids (BCAAs) like leucine, isoleucine, and valine. This does not mean protein is "bad" — adequate protein is essential — but it underscores the importance of dietary balance and avoiding extremes, especially for those already at risk for or diagnosed with insulin resistance.
Q5: What are ceramides, and should I be concerned about them?
Ceramides are fat-derived molecules produced inside muscle cells, especially when you consume large amounts of saturated fats (found in processed meats, many fried foods, and some dairy products). At high levels, ceramides directly block the insulin signaling pathway by inactivating Akt, a critical protein for glucose transport. The good news is that reducing saturated fat intake and increasing exercise can lower intramyocellular ceramide levels over time.
Q6: How do inflammatory kinases like JNK "know" to disrupt insulin signaling?
JNK and IKKβ are stress-response kinases — their job is to respond to signals of cellular danger, like excess fat, inflammatory molecules, or oxidative stress. In this context, they "mean well" — they are trying to protect the cell from overload. But chronically, their activity becomes maladaptive: they phosphorylate IRS-1 at the wrong sites, turning off insulin signaling as a side effect of their protective stress responses. It is a case of the body's defense mechanism causing collateral damage.
Q7: What new treatments are being developed based on these mechanisms?
Several promising directions are being explored: JNK inhibitors (to reduce IRS-1 serine phosphorylation), ceramide synthesis inhibitors (to reduce lipotoxicity), and drugs that enhance PI3K–Akt activity directly. Additionally, the recognition that the gut microbiome influences ceramide and inflammatory cytokine production has opened dietary and probiotic-based intervention strategies. SGLT2 inhibitors and GLP-1 receptor agonists, already in clinical use, may partly exert their metabolic benefits through improving some of these pathways indirectly.
Conclusion: From Molecular Chaos to Metabolic Mastery
Skeletal muscle insulin resistance is not a single "broken switch." As we have explored, it is a sophisticated, multi-level signaling failure. Whether it is the structural "handshake" disruption of IRS-1, the lipotoxic "oil in the gears" from ceramides, or the inflammatory "fire" of the JNK/IKKβ complex, the message is clear: your muscle cells are responding to a high-stress, high-nutrient environment by shutting their doors.
However, the most important takeaway from this molecular map is hope. Because we understand exactly where the relay baton is being dropped, we know exactly how to pick it up. By utilizing the AMPK "bypass road" through physical movement and reducing the BCAA and lipid "traffic jams" through precision nutrition, we can manually reset the cellular machinery.
The laboratory markers like HOMA-IR, Fasting Insulin, and TG/HDL ratio are the early warning signals of this internal signaling failure. By understanding the molecular biology behind those numbers, you are no longer just managing a lab value; you are restoring the fundamental vitality of your cellular life.
Author’s Note
Insulin resistance is often reduced to a laboratory number — fasting glucose, HbA1c, fasting insulin — but the true story unfolds at a far more intricate level. As a physician deeply engaged in metabolic research and clinical medicine, I believe patients deserve to understand what is happening beneath those numbers. When we translate molecular biology into clear, practical language, we move from fear to informed action.
This article was written with two audiences in mind: individuals seeking clarity about their metabolic health, and healthcare professionals who value a concise yet mechanistically accurate synthesis of current evidence. The pathways discussed — IRS-1 signaling, PI3K–Akt disruption, inflammatory kinases, and lipotoxic intermediates — are not abstract biochemical trivia. They represent the real cellular events that precede type 2 diabetes, cardiovascular disease, and metabolic decline.
Importantly, molecular complexity should not be interpreted as inevitability. One of the most empowering insights from modern metabolic science is that these pathways are dynamic. Exercise alters them. Dietary quality alters them. Sleep, stress, body composition, and physical activity patterns all influence them. Skeletal muscle is remarkably adaptable tissue; under the right conditions, it can restore insulin sensitivity to a meaningful degree.
My goal is not to overwhelm with detail, but to replace vague advice with mechanistic understanding. When patients understand why movement improves glucose control or how excess saturated fat influences cellular signaling, lifestyle recommendations become biologically logical rather than abstract rules.
Science is evolving rapidly. As new research refines our understanding of metabolic disease, our approach must evolve with it — grounded in evidence, cautious in interpretation, and always centered on patient empowerment.
Thank you for engaging deeply with this material
Disclaimer: This article is for informational purposes only and does not constitute medical advice. Individual circumstances vary, and treatment decisions should always be made in consultation with qualified healthcare professionals.
Related Articles
Why Skeletal Muscle Is the Body’s Most Powerful Metabolic Organ | DR T S DIDWAL
Obesity and Fatty Liver Disease: What Science Says About Risk and Health | DR T S DIDWAL
Intermittent Fasting: Metabolic Health Benefits and the Evidence on Longevity | DR T S DIDWAL
Activate Your Brown Fat: A New Pathway to Longevity and Metabolic Health | DR T S DIDWAL
References
Dutta, S. (2025). JNK at the helm: decoding obesity-driven insulin resistance. Nature Reviews Endocrinology, 21, 459. https://doi.org/10.1038/s41574-025-01117-9
Li, J., Xu, J., Tian, J., Chen, L., Zhao, L., Yang, Y., Wang, J., Yan, L., Yang, Y., Jiang, Y., Chen, S., Wang, B., Wang, L., & Yang, X. (2026). High-protein diet exacerbates insulin resistance via the JNK/IKKβ-IRS-1 pathway. Endocrinology, Diabetes & Metabolism, 9(1), e70147. https://doi.org/10.1002/edm2.70147
Tang, W., Liu, H., Li, X., Deng, S., & Gao, C. (2026). Influence and treatment of insulin receptor substrate/PI3K/Akt-mediated insulin resistance in diabetes mellitus (Review). Molecular Medicine Reports, 33(2), 63. https://doi.org/10.3892/mmr.2025.13773
Woo, J. R., Bae, S. H., Wales, T. E., Engen, J. R., Lee, J., Jang, H., & Park, S. (2024). The serine phosphorylations in the IRS-1 PIR domain abrogate IRS-1 and IR interaction. Proceedings of the National Academy of Sciences of the United States of America, 121(17), e2401716121. https://doi.org/10.1073/pnas.2401716121