Metainflammation: The Hidden Driver of Premature Vascular Aging and Endothelial Dysfunction
Is your heart aging faster than you think? Discover how endothelial dysfunction drives premature vascular aging and increases early heart disease risk
HEARTMETABOLISM
Dr. T.S. Didwal, M.D.(Internal Medicine)
3/4/202614 min read
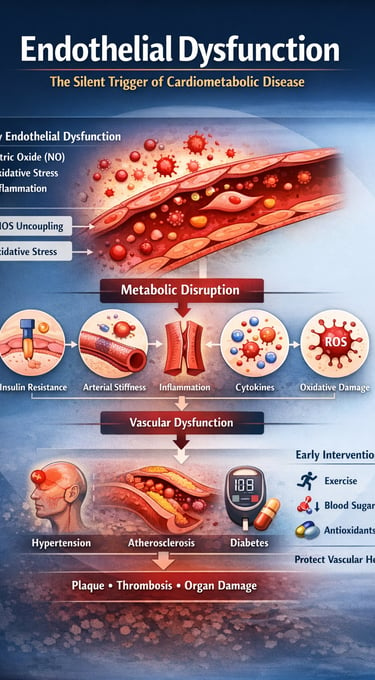
The modern epidemic of obesity, type 2 diabetes, and atherosclerosis shares a quiet, common perpetrator—a state of chronic, low-grade, systemic inflammation so subtle it evades classical detection, yet so pervasive it remodels every organ system it touches. Scientists now define this as metaflammation: a metabolically triggered "smoldering" that persists at a fraction of the intensity of acute infection but inflicts compounding damage over decades. Unlike the self-limiting redness of a wound, metaflammation is non-resolving and omnipresent, driven not by pathogens but by chronic nutrient excess, such as free fatty acids and glucose (Cifuentes et al., 2025).
At the heart of this pathology lies NF-κB, the "master molecular switch." While this transcription factor evolved to coordinate rapid immune defences, under metabolic overload, it becomes a "stuck accelerator," perpetually driving a steady trickle of pro-inflammatory cytokines such as TNF-alpha and IL-6 (Hoffmann et al., 2025). This cytokine relay network creates a vicious feed-forward loop in which inflammation in visceral fat directly impairs insulin signalling in muscle and mitochondrial function in joints (Liu et al., 2026).
The ultimate clinical consequence of this cellular chatter is endothelial dysfunction. The vascular lining, sensitive to these circulating signals, loses its ability to produce nitric oxide, shifts toward a pro-thrombotic state, and becomes permeable to cholesterol (Wang et al., 2026). This transformation serves as the critical bridge through which metabolic stress is translated into cardiovascular catastrophe. By identifying metaflammation as the "common soil" of chronic disease, we move toward a new paradigm of medicine: one in which targeting the underlying inflammatory milieu enables simultaneous prevention of multiple non-communicable diseases.
Metaflammation: The Hidden Driver of the Modern Metabolic Epidemic
The global rise in obesity, Type 2 Diabetes (T2D), and non-alcoholic fatty liver disease (NAFLD) shares a common, silent perpetrator: Metaflammation. Unlike the acute inflammation following an injury, metaflammation is a chronic, low-grade systemic state that remodels organ systems over decades without triggering classic clinical symptoms.
What is Metaflammation? Redefining Chronic Disease
While classical inflammation is a self-limiting defense response (redness, heat, swelling), metaflammation obeys a different set of rules. It is non-resolving and triggered by metabolic substrates rather than pathogens.
Timeline: Measured in years to decades, not days.
Triggers: Excess free fatty acids, glucose, and advanced glycation end-products (AGEs).
Mechanism: Activation of resident immune cells across visceral adipose, liver, and skeletal muscle.
A landmark review by Cifuentes et al. (2025) established metaflammation as the "common soil" linking cardiovascular disease, Alzheimer’s, and even certain cancers. Their research positions chronic inflammation not as a byproduct of metabolic disease, but as its upstream driver.
The Hallmarks of Metaflammation: A Clinical Profile
Metaflammation is defined by a unique set of pathological features that distinguish it from classical, acute immune responses. Understanding these "hallmarks" is essential for identifying patients who may be at risk for systemic metabolic decline before traditional clinical markers flag a problem.
1. Subclinical Cytokine Elevation (The "Smoldering" Fire)
Unlike an acute infection where inflammatory markers skyrocket, metaflammation is characterized by a persistent, low-amplitude signal.
The 2–4× Rule: Pro-inflammatory cytokines remain significantly above baseline but below the standard threshold for acute inflammation diagnosis.
Chronic Signaling: This subtle elevation is sufficient to disrupt cellular signaling, particularly insulin sensitivity, over years of exposure.
2. Multi-Organ Activation Sites
Metaflammation is not localized; it activates resident immune cells across the body’s most metabolically active tissues:
Visceral Adipose Tissue: The primary "endocrine" engine of inflammation.
Vascular Endothelium: The critical bridge to cardiovascular disease.
The Liver & Skeletal Muscle: Driving systemic insulin resistance and metabolic inflexibility.
The Hypothalamus: Potentially altering central appetite and energy regulation.
3. The Molecular Fingerprint (Key Mediators)
The "cytokine relay" of metaflammation involves a specific repertoire of proteins that drive tissue remodeling and insulin resistance:
Primary Cytokines: TNF-alpha, IL-6, and IL-1beta
Adipokines & Chemokines: Resistin and $MCP-1$ (which recruits macrophages into fat tissue).
Systemic Markers: High-sensitivity C-reactive protein (hsCRP).
4. Upstream Drivers: Nutrient & Cellular Stress
Metaflammation is triggered by metabolic excess rather than external pathogens.
Lipotoxicity & Glucotoxicity: Excess saturated fatty acids and glucose act as direct ligands for inflammatory receptors.
Gut Dysbiosis: "Leaky gut" allows bacterial toxins to enter the blood, fueling systemic tone.
Intracellular Stress: Endoplasmic reticulum (ER) stress and cellular hypoxia in expanding fat tissue ignite the NF-κB match.
5. The Self-Perpetuating Feed-Forward Loop
Perhaps the most dangerous hallmark of metaflammation is its ability to sustain itself.
Adipocyte Dysfunction: Inflammatory mediators cause fat cells to become further dysfunctional, releasing more fatty acids and cytokines.
The Loop: This creates a vicious cycle where inflammation drives metabolic failure, and metabolic failure further fuels inflammation.
NF-κB: The “Master Molecular Switch” Behind Metaflammation
If chronic metabolic inflammation (metaflammation) is a slow-burning fire inside the body, NF-κB is the match that keeps relighting it.
NF-κB (Nuclear Factor kappa-light-chain-enhancer of activated B cells) is a powerful protein complex that controls inflammation at the genetic level. It acts like a master switch — turning on hundreds of inflammatory genes.
What Exactly Is NF-κB?
A family of five related proteins (RelA/p65, RelB, c-Rel, p50, p52)
Found inside almost every cell in the body
Controls the production of inflammatory chemicals such as:
TNF-α
IL-6
IL-1β
Adhesion molecules (ICAM-1, VCAM-1)
In simple terms: NF-κB decides how loudly your immune system speaks.
Why NF-κB Is Essential — and Also Dangerous
NF-κB has a dual role:
When Working Normally
Fights infections
Coordinates immune defense
Helps wounds heal
Protects against pathogens
❌ When Chronically Activated
Drives low-grade inflammation
Promotes insulin resistance
Damages blood vessels
Contributes to heart disease, diabetes, and neurodegeneration
In metabolic disease, NF-κB doesn’t shut off properly.
How NF-κB Gets Activated in Obesity and Metabolic Disease
There are two main pathways:
The Canonical (Classical) Pathway
Triggered by:
Excess saturated fats (like palmitate)
High blood sugar
Gut-derived endotoxins entering circulation
Chronic visceral fat expansion
What happens:
Receptors like TLR4 detect metabolic stress.
An enzyme complex (IKK) activates NF-κB.
NF-κB moves into the nucleus.
Inflammatory genes switch on.
Result:
Increased TNF-α and IL-6
Vessel wall inflammation
Reduced nitric oxide production
Early endothelial dysfunction
2️⃣ The Non-Canonical Pathway
Activated by immune remodeling signals in chronic obesity.
This pathway:
Alters immune cell behavior in fat tissue
Turns adipose tissue into a pro-inflammatory endocrine organ
Sustains long-term metaflammation
Why This Matters for Your Arteries
When NF-κB stays mildly but persistently active:
Blood vessels lose flexibility
Nitric oxide production falls
Adhesion molecules increase
Plaque formation risk rises
You may feel completely normal — but vascular aging has already begun.
The “Stuck Accelerator” Concept
The same NF-κB system that evolved to fight infections is now overstimulated by:
Too much sugar
Too much saturated fat
Chronic stress
Visceral obesity
Instead of turning on briefly and shutting off, it remains partially active for years.
It doesn’t create dramatic symptoms.
It creates slow tissue damage over decades.
Clinical Takeaway
NF-κB is not inherently harmful. It is essential for survival.
But in modern metabolic overload, it becomes chronically activated — sustaining the low-grade inflammation that drives:
Premature vascular aging
Insulin resistance
Atherosclerosis
Fatty liver disease
Neuroinflammation
Reducing metabolic stress — through weight control, exercise, fibre-rich nutrition, sleep optimization, and glucose stability — helps quiet this molecular switch.
NF-κB is the molecular bridge between metabolic overload and chronic inflammatory disease.
When we calm the switch, we calm the fire.
Cytokine Cross-Talk: The Inflammatory Relay Network Driving Metaflammation
Metaflammation is not caused by one inflammatory molecule.
It is driven by a cytokine relay network — a coordinated cross-talk system where inflammatory messengers activate, amplify, and sustain each other across multiple organs.
This is how a local problem with belly fat can eventually damage:
The heart
The liver
The joints
The brain
What Is Cytokine Cross-Talk?
Cytokine cross-talk refers to the interaction between inflammatory signaling molecules that:
Amplify each other’s production
Activate shared pathways like NF-κB
Spread inflammation across tissues
Disrupt metabolism and vascular function
In simple terms: Inflammation talks to itself — and the conversation spreads.
The Key Players in the Cytokine Relay Network
1️⃣ TNF-α: The Feed-Forward Amplifier
Tumor Necrosis Factor-alpha (TNF-α) is a central pro-inflammatory cytokine.
It:
Activates NF-κB in target cells
Stimulates more TNF-α production
Induces IL-6 and IL-1β release
Impairs insulin signaling
Promotes endothelial dysfunction
This creates a self-sustaining inflammatory loop.
2️⃣ IL-6: From Exercise Helper to Metabolic Harm
Interleukin-6 (IL-6) has a dual identity.
During Exercise
Acts as a beneficial myokine
Supports glucose regulation
Improves fat metabolism
In Chronic Obesity
When continuously released from:
Visceral adipocytes
Liver Kupffer cells
It:
Activates JAK/STAT3 signaling
Raises C-reactive protein (CRP)
Promotes insulin resistance
Drives systemic inflammation
Chronic elevation of IL-6 becomes a cardiometabolic risk amplifier.
Cytokine Cross-Talk Extends Beyond the Heart
Inflammation–metabolism crosstalk affects multiple systems.
A 2026 study by Liu et al. showed that in osteoarthritis:
NF-κB drives IL-6 and IL-17 overproduction
Cells lose mitochondrial efficiency
Energy production declines
Matrix breakdown accelerates
This proves that cytokine cross-talk:
Disrupts bioenergetics
Impairs cellular metabolism
Extends beyond immune cells
Drives systemic metabolic aging
Metaflammation is a whole-body network disorder.
The Adipokine Imbalance: When Fat Becomes an Endocrine Organ
In metabolic disease, visceral fat changes its hormonal output.
This shift is called adipokine imbalance.
What Happens in Healthy Fat Tissue
High adiponectin (anti-inflammatory)
Improved insulin sensitivity
Suppressed NF-κB activity
Protected blood vessels
What Happens in Metaflammation
Anti-inflammatory signals drop:
↓ Adiponectin
Pro-inflammatory signals rise:
↑ Leptin
↑ Resistin
↑ Chemerin
↑ Visfatin
Hypertrophied fat cells release MCP-1 (CCL2), which:
Recruits immune cells
Converts macrophages into inflammatory M1 type
Produces more TNF-α and IL-6
This creates a self-amplifying adipose inflammatory loop.
Why This Matters for Cardiovascular Risk
This adipose-centred cytokine relay:
Spreads inflammation systemically
Impairs insulin signaling
Reduces nitric oxide in arteries
Increases endothelial adhesion molecules
Accelerates premature vascular aging
Visceral fat becomes a molecular gateway to heart disease.
Endothelial Dysfunction: The Critical Cardiovascular Bridge
The vascular endothelium is a single-cell layer lining nearly 60,000 miles of blood vessels.
It is not just a passive barrier — it is a dynamic endocrine organ that regulates:
Blood flow
Inflammation
Clotting
Vascular tone
Immune cell trafficking
In metaflammation, this delicate lining becomes both:
A primary target of inflammatory cytokines
An amplifier of systemic inflammation
Endothelial dysfunction is now recognized as the critical mechanistic bridge linking chronic inflammation to:
Atherosclerosis
Hypertension
Insulin resistance
Acute cardiovascular events
What Is Endothelial Dysfunction?
Endothelial dysfunction refers to a shift from a protective state to a pro-inflammatory, pro-thrombotic, pro-atherogenic phenotype.
It is characterized by:
↓ Nitric oxide (NO) availability
↑ Adhesion molecule expression
↑ Oxidative stress
↑ Vascular permeability
↑ Clotting tendency
Importantly, it begins years before detectable plaque formation.
Four Core Pathways of Endothelial Injury in Metaflammation
1️⃣ eNOS Uncoupling: Loss of Nitric Oxide Protection
Nitric oxide (NO) keeps arteries:
Relaxed
Flexible
Anti-inflammatory
Anti-thrombotic
In metaflammation:
Oxidative stress depletes tetrahydrobiopterin (BH4)
Endothelial nitric oxide synthase (eNOS) becomes “uncoupled”
Instead of producing nitric oxide, it produces superoxide
Result:
Loss of vasoprotection
Increased reactive oxygen species
Accelerated vascular aging
2️⃣ Adhesion Molecule Overexpression
Chronic NF-κB activation in endothelial cells increases:
ICAM-1
VCAM-1
E-selectin
These molecules:
Allow monocytes to stick to vessel walls
Promote immune cell infiltration
Initiate atherosclerotic plaque formation
This is the first cellular step in atherogenesis.
3️⃣ Barrier Disruption and LDL Leakage
Inflammatory cytokines such as TNF-α and IL-1β:
Disrupt tight junction proteins (claudin, occludin, VE-cadherin)
Increase endothelial permeability
This allows:
LDL particles to enter the vessel wall
Oxidized cholesterol deposition
Foam cell formation
The result: structural plaque development.
4️⃣ Pro-Thrombotic Shift
In dysfunctional endothelium:
Thrombomodulin decreases
PAI-1 increases
von Willebrand factor rises
The vascular environment shifts toward:
Increased clot formation
Elevated acute coronary event risk
Greater stroke vulnerability
Endothelial Dysfunction Precedes Plaque
Research shows endothelial dysfunction appears years before visible atherosclerosis.
This makes it:
An early biomarker of cardiovascular risk
A reversible therapeutic target
Clinical tools include:
Flow-Mediated Dilation (FMD)
Serum ICAM-1 and VCAM-1 levels
hsCRP as a systemic inflammation proxy
Early detection = early intervention.
Metaflammation: The “Common Soil” of Chronic Disease
Low-grade chronic inflammation acts as a shared biological substrate across non-communicable diseases.
Elevated hsCRP consistently predicts:
Type 2 diabetes
Major adverse cardiac events
Colorectal cancer
Alzheimer’s disease
This suggests a single inflammatory environment can generate multiple disease outcomes depending on organ vulnerability.
Metaflammation: The Future of Pleiotropic Anti-Inflammatory Therapy
If metaflammation is the "common soil" of chronic disease, then the future of medicine lies in pleiotropic anti-inflammatory interventions. Emerging research from 2025–2026 suggests that by targeting the core molecular machinery of metabolically triggered inflammation, we can simultaneously treat obesity, cardiovascular disease, and neurodegeneration.
Pharmacological Frontiers: Disrupting the NF-κB Match
Since NF-κB acts as the master molecular switch for metaflammation, pharmacological research is shifting toward high-precision inhibition.
1. Advanced NF-κB Pathway Inhibitors
Mechanisms: Current research explores IKK-β inhibitors, proteasome inhibitors, and HDAC inhibitors to modulate transcriptional activity.
The Nanoparticle Breakthrough: To avoid systemic immunosuppression, nanoparticle-encapsulated IKK inhibitors are being developed to target high-risk tissues, such as visceral adipose tissue and the vascular endothelium, directly.
2. Repurposed Metabolic "Gems"
Metformin: Beyond glucose control, Metformin inhibits NF-κB and the NLRP3 inflammasome, acting as a potent anti-meta-inflammatory agent.
Statins: Their cardiovascular benefits are now understood to be partly independent of LDL-lowering, driven instead by direct NF-κB suppression.
3. Biological Cytokine Blockade
Validated by the CANTOS trial, targeting specific cytokines like IL-1β and IL-6 provides clinical proof that reducing the inflammatory milieu directly prevents major adverse cardiac events (MACE).
Lifestyle Interventions:
Lifestyle remains the most accessible and potent tool for reducing meta-inflammatory tone.
Caloric Restriction: Rapidly reduces visceral fat mass and restores levels of the "vessel-protective" hormone, adiponectin.
The Mediterranean Diet: High in polyphenols, omega-3s, and fermentable fiber, this pattern consistently lowers biomarkers like hsCRP and ICAM-1.
Aerobic Exercise: Activates adenylate kinase and suppresses NF-κB signaling in skeletal muscle, functioning as "mechanical medicine" for the endothelium.
Summary of the landmark research
1. The "Common Soil" Theory (Cifuentes et al., 2025)
Core Finding: Low-grade chronic inflammation is established as the primary upstream driver—rather than a consequence—of major non-communicable diseases (NCDs).
The "Unified Substrate": Identifies a singular pathophysiological thread connecting obesity, Type 2 Diabetes, cancer, and Alzheimer’s disease.
Clinical Pearl: Shifting the treatment focus toward this "common soil" allows for the simultaneous reduction of risk across multiple disease categories.
2. NF-κB: The Master Molecular Switch (Hoffmann et al., 2025)
Core Finding: Defines the "dual identity" of NF-κB; while essential for acute immune defense, it becomes a "stuck accelerator" in the presence of chronic nutrient excess.
Mechanistic Insight: Highlights the non-canonical pathway as a major contributor to how visceral fat transforms into a pro-inflammatory endocrine organ.
Strategic Takeaway: The future of therapy lies in tissue-selective inhibition (targeting the "stuck accelerator") rather than systemic blockade, which could compromise the immune system.
3. The Inflammation-Metabolism Crosstalk (Liu et al., 2026)
Core Finding: Using osteoarthritis as a model, researchers discovered that chronic cytokine exposure (IL-6, IL-17) directly impairs mitochondrial bioenergetics.
The Vicious Cycle: Inflammatory signals cause cells to produce more cytokines while simultaneously losing the ability to generate energy efficiently.
Therapeutic Potential: Identifies novel targets like AMPK activators and sirtuin modulators as ways to "reboot" cellular metabolism and quench inflammation.
4. The Endothelial Bridge (Wang et al., 2026)
Core Finding: Confirms the vascular endothelium as the critical bridge where systemic metaflammation is translated into physical cardiovascular damage.
Four-Path Breakdown: Identifies eNOS uncoupling, adhesion molecule overexpression, barrier disruption, and pro-thrombotic shifts as the four cardinal drivers of decay.
Diagnostic Value: Since endothelial dysfunction precedes arterial plaque by years, markers like Flow-Mediated Dilation (FMD) and soluble adhesion molecules (ICAM-1) are established as the best early-warning tools.
Metaflammation: Summary
1️⃣ Definition and Core Concept
Metaflammation (metabolic inflammation) is a chronic, low-grade, systemic inflammatory state driven by nutrient excess and metabolic stress.
Unlike acute inflammation (short-term and protective), metaflammation is persistent, silent, and maladaptive.
It serves as a common mechanistic link between obesity, insulin resistance, type 2 diabetes, atherosclerosis, sarcopenia, and premature vascular aging.
2️⃣ Molecular Triggers
Chronic overnutrition and visceral adiposity initiate inflammatory signaling.
Enlarged adipocytes release pro-inflammatory cytokines (TNF-α, IL-6, MCP-1).
These signals recruit immune cells, particularly M1 macrophages, into adipose tissue.
Key inflammatory pathways activated include:
NF-κB
JNK
NLRP3 inflammasome
Mitochondrial dysfunction and lipotoxicity amplify oxidative stress.
3️⃣ Impact on Insulin and Metabolism
Inflammatory mediators impair IRS-1 phosphorylation, disrupting PI3K-Akt signaling.
Result: systemic insulin resistance.
Skeletal muscle shows reduced glucose uptake and mitochondrial decline.
The liver develops steatosis and contributes to dysglycemia.
Pancreatic beta cells experience chronic stress and functional decline.
4️⃣ Vascular Consequences
Endothelial cells exhibit reduced nitric oxide bioavailability.
Oxidative stress increases vascular stiffness.
Early endothelial dysfunction emerges — a precursor to atherosclerosis.
Metaflammation contributes to premature vascular aging.
5️⃣ System-Wide Extension
Not limited to adipose tissue.
Extends to:
Liver (NAFLD)
Muscle (metabolic inflexibility)
Pancreas (beta-cell failure)
Brain (neuroinflammation)
Represents a network disorder, not a single-organ disease.
6️⃣ Aging and Cellular Effects
Promotes cellular senescence.
Accelerates telomere shortening.
Drives epigenetic alterations.
Contributes to sarcopenia and biological aging acceleration.
7️⃣ Clinical Implications
Shifts focus beyond glucose control alone.
Evidence-based anti-inflammatory strategies include:
Structured resistance and aerobic exercise
Caloric modulation and high-fiber diets
Omega-3 intake
Sleep optimization and stress reduction
Certain medications (GLP-1 receptor agonists, SGLT2 inhibitors) demonstrate secondary anti-inflammatory effects.
Core Takeaway
Metaflammation is the common inflammatory soil underlying modern cardiometabolic disease. Early intervention targeting metabolic inflammation may redefine prevention and slow vascular and systemic decline.
Frequently Asked Questions (FAQs)
What exactly is metaflammation and how is it different from regular inflammation?
Metaflammation is a state of chronic, low-grade, systemic inflammation driven by metabolic overload — excess lipids, glucose, and their metabolic by-products — rather than pathogens or injury. Unlike classical acute inflammation, which resolves in days and is characterised by visible redness and swelling, metaflammation operates at subclinical intensity (cytokines elevated 2–4× above baseline), does not resolve spontaneously, and persists for years to decades. It is detectable through biomarkers like high-sensitivity CRP (hsCRP), IL-6, and soluble adhesion molecules, rather than clinical signs of acute inflammation (Cifuentes et al., 2025).
How does NF-κB activation specifically contribute to cardiovascular disease risk?
NF-κB, when chronically activated in endothelial cells and vascular smooth muscle cells, drives transcription of adhesion molecules (ICAM-1, VCAM-1, E-selectin), pro-inflammatory cytokines (TNF-α, IL-6), and pro-thrombotic factors. These collectively promote monocyte adhesion and transmigration into the arterial wall, LDL oxidation and foam cell formation, plaque development, and eventually plaque rupture leading to acute myocardial infarction or stroke. NF-κB also activates matrix metalloproteinases that degrade the fibrous plaque cap, increasing its vulnerability to rupture (Hoffmann et al., 2025; Wang et al., 2026).
What biomarkers can I use to measure metaflammation clinically?
High-sensitivity CRP (hsCRP) is the most clinically validated and widely available biomarker, with levels below 1 mg/L considered low risk, 1–3 mg/L intermediate, and above 3 mg/L high risk. Other useful markers include IL-6, TNF-α, fibrinogen, soluble ICAM-1, soluble VCAM-1, white blood cell count, and ferritin. For vascular-specific endothelial dysfunction, brachial artery flow-mediated dilation (FMD) is the gold-standard non-invasive measure, while circulating endothelial microparticles and asymmetric dimethylarginine (ADMA) provide additional granularity (Wang et al., 2026; Cifuentes et al., 2025).
Can metaflammation be reversed through lifestyle changes alone?
Yes, substantial evidence supports that lifestyle intervention can meaningfully reduce metaflammatory tone. Weight loss of 5–10% of body mass significantly reduces hsCRP, IL-6, TNF-α, and ICAM-1 levels, with some studies showing normalisation of endothelial function within 8–12 weeks of caloric restriction. Aerobic exercise has independent anti-inflammatory effects via AMPK activation, myokine release, and visceral fat reduction. The Mediterranean dietary pattern reduces hsCRP by approximately 20–30% in randomised controlled trials. However, genetic factors, gut microbiome composition, and degree of metabolic dysregulation influence the magnitude of response.
Why is endothelial dysfunction described as the "critical cardiovascular bridge" in metaflammation?
The endothelium is uniquely positioned at the intersection of systemic inflammatory signalling and cardiovascular pathology. As a primary target of circulating cytokines — TNF-α, IL-1β, and IL-6 all directly impair endothelial nitric oxide synthase (eNOS) function — the endothelium translates systemic metaflammation into local vascular pathology through eNOS uncoupling, adhesion molecule expression, barrier disruption, and pro-thrombotic activation. Because endothelial dysfunction precedes detectable atherosclerosis by years, it represents both an early warning signal of metaflammatory burden and a therapeutic window for cardiovascular prevention before irreversible structural damage occurs (Wang et al., 2026).
How does cytokine cross-talk amplify metaflammation beyond the initial metabolic trigger?
Cytokine cross-talk creates self-amplifying inflammatory cascades: TNF-α activates NF-κB in target cells, generating more TNF-α, IL-6, and IL-1β. IL-6 signals through JAK/STAT3 to produce acute-phase reactants and further inflammatory mediators. IL-1β, processed by the NLRP3 inflammasome (itself activated by cholesterol crystals and fatty acids), drives additional NF-κB activation in macrophages and endothelial cells. MCP-1 recruits circulating monocytes to adipose and vascular tissue, where they differentiate into pro-inflammatory M1 macrophages and amplify local cytokine production. This multi-node relay network means that a localised metabolic insult — say, visceral adipose expansion — rapidly generates systemic inflammatory consequences (Liu et al., 2026; Cifuentes et al., 2025).
What are the most promising pharmacological strategies currently being developed to target metaflammation?
Several converging therapeutic strategies show translational promise. IL-1β blockade (canakinumab), proven in the CANTOS trial, remains the most clinically validated approach. NLRP3 inflammasome inhibitors (e.g., colchicine, MCC950 analogues) are in active clinical trials for cardiovascular endpoints. Selective NF-κB pathway inhibitors delivered via adipose-targeted nanoparticles aim to suppress metaflammation without systemic immunosuppression. SGLT2 inhibitors and GLP-1 receptor agonists, beyond their glycaemic effects, demonstrate significant reductions in hsCRP, IL-6, and ICAM-1 in clinical trials. Metformin's AMPK-activating, NF-κB-suppressing properties are being investigated for cancer and neurodegenerative indications based on the metaflammation paradigm (Hoffmann et al., 2025; Liu et al., 2026).
Author’s Note
Endothelial dysfunction represents one of the most clinically underappreciated turning points in cardiometabolic disease. In daily practice, we often encounter hypertension, diabetes, or atherosclerosis only after structural vascular damage has become measurable. Yet beneath these diagnoses lies a deeper disturbance — impaired endothelial signaling.
This article was written to reframe vascular disease not as a late-stage plaque problem, but as an early signaling disorder characterized by reduced nitric oxide bioavailability, oxidative stress amplification, inflammatory activation, and microvascular instability. The vascular endothelium is not merely a passive lining of blood vessels; it is a dynamic endocrine interface integrating metabolic, hemodynamic, and immune signals.
From a clinical perspective, this shift matters. When we begin to view cardiometabolic disorders through the lens of endothelial biology, prevention becomes more precise. Exercise is no longer just “lifestyle advice,” but a shear-stress–mediated enhancer of nitric oxide synthesis. Glycemic control becomes a strategy to limit oxidative endothelial injury. Weight reduction reduces inflammatory cytokine burden at the vascular interface. Blood pressure control becomes preservation of endothelial integrity.
The intent here is not to replace established cardiovascular guidelines, but to deepen the physiological understanding that underpins them. By identifying endothelial dysfunction early — before irreversible vascular remodelling — clinicians can intervene at the level of mechanism rather than at the level of consequence.
As metabolic medicine advances, preserving endothelial function may prove to be one of the most powerful unifying strategies linking diabetes care, obesity management, hypertension treatment, and cardiovascular prevention.
Understanding the endothelium is understanding the earliest opportunity for vascular protection.
Disclaimer: This article is for informational purposes only and does not constitute medical advice. Individual circumstances vary, and treatment decisions should always be made in consultation with qualified healthcare professionals.
Related Articles
Tired All Day, Awake at Night? The Redox and Mitochondrial Link | DR T S DIDWAL
Obesity and Fatty Liver Disease: What Science Says About Risk and Health | DR T S DIDWAL
Intermittent Fasting: Metabolic Health Benefits and the Evidence on Longevity | DR T S DIDWAL
Activate Your Brown Fat: A New Pathway to Longevity and Metabolic Health | DR T S DIDWAL
References
Cifuentes, M., Verdejo, H. E., Castro, P. F., Corvalan, A. H., Ferreccio, C., Quest, A. F. G., Kogan, M. J., & Lavandero, S. (2025). Low-grade chronic inflammation: A shared mechanism for chronic diseases. Physiology, 40(1), 4–25. https://doi.org/10.1152/physiol.00021.2024
Hoffmann, A., Cheng, G., & Baltimore, D. (2025). NF-κB: Master regulator of cellular responses in health and disease. Immunity & Inflammation, 1, 2. https://doi.org/10.1007/s44466-025-00014-0
Liu, B., Zhang, M., Gao, L., & Yang, H. (2026). Targeting inflammation-metabolism crosstalk: Current status and challenges of novel therapeutic strategies for osteoarthritis. Frontiers in Medicine, 12, 1754693. https://doi.org/10.3389/fmed.2025.1754693
Wang, Z., Yang, Y., Wang, Q., Wang, L., Zhao, Y., Qian, X., Feng, R., & Qian, J. (2026). Pathological mechanisms and clinical research progress of endothelial dysfunction. Frontiers in Cardiovascular Medicine, 13, 1749548. https://doi.org/10.3389/fcvm.2026.1749548