Why Your Metabolism Slows Down: The Hidden Role of Mitochondrial Dysfunction in Obesity
Discover how mitochondrial dysfunction drives obesity, insulin resistance, and metabolic disease — plus evidence-based strategies to restore your cellular energy system and metabolism.
OBESITY
Dr. T.S. Didwal, M.D.(Internal Medicine)
6/13/202623 min read
How Does Mitochondrial Dysfunction Impact Obesity?
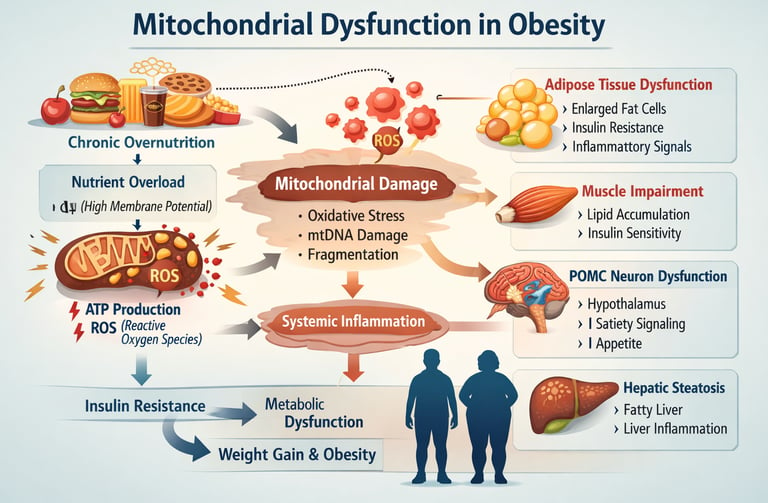
Mitochondrial dysfunction shifts obesity from a basic caloric imbalance to a complex cellular bioenergetic failure. Chronic overnutrition elevates mitochondrial membrane potential, inducing electron leaks that accelerate reactive oxygen species (ROS) production. This pathologically impairs ATP synthesis efficiency, causing systemic cellular energy insufficiency, chronic fatigue, and metabolic inflexibility
Key Takeaways: Mitochondrial Dysfunction & Obesity
1. Cellular Bioenergetic Failure vs. Caloric Excess
The Biomedical Mechanism: Obesity is fundamentally a disorder of cellular bioenergetics rather than a simple caloric imbalance. Chronic overnutrition elevates mitochondrial membrane potential, inducing an electron leak from the electron transport chain (ETC) and accelerating the production of reactive oxygen species (ROS). This pathology reduces ATP synthesis efficiency despite substrate abundance, forcing a state of intracellular energy insufficiency.
The Patient Reality: It is common to blame chronic fatigue on laziness or a lack of willpower. In reality, the body's cells are having a hard time turning stored fat into actual energy. This creates a frustrating situation where a person has plenty of stored "fuel" on their body, but their cells are essentially running on empty, causing real, physical exhaustion.
2. The Core Triad of Organelle Deficits
The Biomedical Mechanism: Mitochondrial decay in obese phenotypes manifests through three distinct axes: impaired biogenesis, defective mitophagy, and pathological fission. Obesity suppresses PGC-1alpha signaling, downregulating the creation of new mitochondria. Concurrently, the impairment of PINK1–Parkin-mediated mitophagy allows damaged, toxic organelles to accumulate, while RalA-driven DRP1 activation forces fragmentation, reducing overall oxidative capacity.
The Patient Reality: Think of this like an assembly line breakdown inside the body: the cells have stopped making new, healthy batteries, they have lost the ability to throw away the old, broken ones, and the batteries that are left are cracked and weak. This explains why exercise can feel almost impossible—the body's internal engines are physically broken down.
3. Spatial and Tissue-Specific Pathophysiology
The Biomedical Mechanism: Mitochondrial defects are highly tissue-specific, driving distinct downstream pathologies. Visceral adipose tissue demonstrates starker reductions in oxidative capacity and higher ROS emission than subcutaneous fat. In skeletal muscle, incomplete fatty acid oxidation causes the accumulation of lipotoxic intermediates (ceramides and diacylglycerols), which directly inhibit insulin signaling (IRS-1/GLUT4). In the hypothalamus, mitochondrial impairment within POMC neurons disrupts central satiety signaling.
The Patient Reality: Weight struggles are not just about a number on the scale; different parts of the body are struggling in different ways. For instance, when a person can't seem to ever feel full, it is often because the "I'm full" switch in the brain isn't getting enough power to work. Similarly, muscle cells can get clogged up with cellular "exhaust fumes," making it harder for the body to manage blood sugar.
4. mtDNA Leakage and the Chronic Inflammatory Cascade
The Biomedical Mechanism: When damaged mitochondria fragment and rupture, they release mitochondrial DNA (mtDNA) into the cytoplasm. Because mtDNA retains bacterial characteristics, it acts as a damage-associated molecular pattern (DAMP), triggering the NLRP3 inflammasome and cGAS-STING pathways. This elevates circulating pro-inflammatory cytokines which feed back to further suppress PGC-1alpha, locking the patient into a chronic, self-reinforcing systemic inflammatory loop.
The Patient Reality: This cellular damage helps explain why weight struggles are often accompanied by issues like aching joints and chronic "brain fog." When the tiny power plants inside cells get damaged, they can spill their internal contents into the body. The immune system mistakes this spilled material for an infection, which sets off a wave of body-wide inflammation.
5. The Substrate Overload Paradox
The Biomedical Mechanism: The "overload paradox" dictates that an oversupply of metabolic substrate directly diminishes net energy output. Hypernutrition forces an influx of electrons into the ETC, pushing membrane potential beyond the optimal threshold required for ATP synthase. This stalls the chain and causes premature electron reduction of molecular oxygen to superoxide. The net calculation yields a higher caloric intake resulting in elevated oxidative stress and a severely diminished ATP-to-ROS ratio.
The Patient Reality: The old advice to "eat less and move more" can backfire when a person's metabolism is overwhelmed. When cells are constantly flooded with too much food, the internal machinery gets choked and jammed. Paradoxically, taking in more food actually causes the body's power plants to stall, leaving the person with less day-to-day energy.
6. Exercise as a Corrective Therapeutic Stimulus
The Biomedical Mechanism: Physical exercise serves as the most reliable non-pharmacological stimulus for mitochondrial quality control. Contraction-induced cellular stress upregulates PGC-1alpha, expanding the mtDNA copy number and restoring ETC complex expression. Specifically, low-intensity continuous exercise (Zone 2) optimizes lipid oxidation capacity, while progressive resistance training increases skeletal muscle mitochondrial density and restores insulin receptor sensitivity.
The Patient Reality: This completely changes how we look at exercise. It isn't a punishment to "burn off calories" or earn food. Instead, moving the body acts like a direct repair order. Easy, steady cardio (like a brisk walk) and strength training are tools used to patch up, rebuild, and add more power plants to the body's internal grid.
7. Metabolic Inflexibility and Substrate Lock
The Biomedical Mechanism: Healthy metabolic systems display high substrate flexibility, seamlessly switching between carbohydrate and lipid oxidation based on nutritional status and endocrine signals. In obese states, mitochondrial fragmentation, ETC enzymatic dysfunction paralyze this transition network. This results in severe postprandial (post-meal) hyperglycemia alongside an inability to mobilize and oxidize endogenous lipids during periods of fasting.
The Patient Reality: A healthy metabolism can easily switch between burning sugar from meals and burning stored body fat for fuel. But when these internal systems are damaged, the body loses that flexibility and gets stuck in one gear. This is why someone might feel completely shaky, starving, and dizzy just two hours after eating, while still finding it incredibly difficult to lose fat when they skip a meal.
8. Lifespan Plasticity and Epigenetic Reversibility
The Biomedical Mechanism: Mitochondrial networks retain profound plasticity and are highly modifiable across the human lifespan. Targeted clinical interventions—including structured time-restricted feeding, deep sleep optimization, and thermal hormesis (sauna/cold stress)—activate the nutrient-sensing AMPK and SIRT1 pathways. This double-pronged activation accelerates both mitophagy of degraded organelles and the biogenesis of healthy ones, showing measurable improvements in mtDNA copy number and oxidative phosphorylation within an 8-to-12-week window.
The Patient Reality: Years or even decades of dealing with weight and fatigue do not mean the metabolism is permanently broken. These tiny cellular power plants are incredibly adaptable. By giving the body the right signals through consistent habits—like protecting sleep, spacing out meals, and using exercise—the body can completely remodel its energy system in as little as two to three months.
1. Why Obesity Is a Cellular Energy Crisis
Most people think of obesity as a problem of eating too much and moving too little. But emerging science tells a more complex — and more hopeful — story.
Obesity begins not at the dinner table, but inside your cells.
At the heart of this story are your mitochondria — tiny organelles responsible for converting the food you eat into usable energy. When these cellular engines malfunction, the downstream consequences are profound: fat accumulates faster, insulin stops working properly, hunger signals go haywire, and weight loss becomes biologically — not just behaviorally — difficult.
A growing body of research from 2024–2026 now frames obesity as a disease of mitochondrial dysfunction rather than simple caloric excess. Understanding this shift changes everything about how we approach weight management, metabolic health, and long-term disease prevention.
In this article, you will learn:
Exactly how mitochondria produce energy — and how that process breaks down in obesity
Why do excess calories paradoxically reduce your body's ability to generate usable energy
How mitochondrial dysfunction affects fat tissue, muscle, liver, and even the brain
The most powerful, evidence-based strategies to restore cellular energy and reverse metabolic disease
2. How Mitochondria Power Your Metabolism
Mitochondria are far more than "powerhouses." They are the metabolic command centers of your cells — integrating nutrient signals, managing oxidative stress, and determining whether your body stores energy or burns it.
The Engine: Oxidative Phosphorylation (OXPHOS)
Your cells generate ATP — the universal energy currency — through a process called oxidative phosphorylation (OXPHOS). During this process:
Electrons derived from food (glucose, fatty acids) flow through the electron transport chain (ETC) — a series of protein complexes embedded in the inner mitochondrial membrane
As electrons move through the chain, protons are pumped across the membrane, creating an electrochemical gradient called the proton motive force (Δp)
This gradient drives ATP synthase, producing ATP
The efficiency of this system is captured in one equation:
Δp = Δψ − 60·ΔpH
Where Δψ is the membrane potential (electrical gradient) and ΔpH is the proton concentration difference
In a healthy cell, this engine runs cleanly and efficiently. In obesity, it does not.
The Obesity Connection: When More Fuel Means Less Energy
Here is the paradox that defines modern metabolic disease:
Excess calories do not produce more energy — they reduce energy efficiency.
When mitochondria are flooded with fuel, the membrane potential (Δψ) rises beyond its optimal range. Electrons begin to "leak" out of the transport chain and react directly with oxygen to form reactive oxygen species (ROS) — highly reactive molecules that damage cellular structures.
The result: instead of efficient ATP production, you get oxidative stress, damaged DNA, and impaired metabolism.
3. What Is Mitochondrial Dysfunction? A 3-Pillar Framework
Mitochondrial dysfunction is not a single defect. It is a multidimensional breakdown across three interconnected systems:
Pillar 1: Impaired Biogenesis (Your Body Stops Building New Mitochondria)
The protein PGC-1α acts as the master switch for mitochondrial production. In obesity, chronic inflammation and oxidative stress suppress PGC-1α activity. The result:
Fewer mitochondria are made
Energy capacity declines
Metabolic flexibility is lost — the ability to switch between burning carbohydrates and fat
Population studies confirm this: lower mitochondrial DNA copy number (a direct marker of biogenesis) is strongly associated with increased cardiometabolic risk.
Pillar 2: Impaired Mitophagy (The Recycling System Breaks Down)
Healthy cells continuously identify and destroy damaged mitochondria through mitophagy — a quality-control process guided by the PINK1–Parkin protein pathway.
In obesity, this system becomes overwhelmed. Damaged mitochondria accumulate, continuing to leak ROS and releasing their contents into the cell — triggering further inflammation and cellular damage.
Pillar 3: Altered Dynamics (Mitochondria Fragment Into Inefficient Pieces)
Healthy mitochondria form elongated, interconnected networks that maximize energy output. In obesity, this balance shifts dramatically toward fission — the splitting of mitochondria into small, fragmented units.
A landmark 2024 study published in Nature Metabolism identified the protein RalA as a key driver of this fragmentation in white adipose tissue. When RalA activates DRP1 (the primary fission protein), mitochondria break apart into smaller, less efficient organelles that:
Produce less ATP
Generate more ROS
Are harder to recycle via mitophagy
4. How Chronic Overnutrition Damages Your Mitochondria
The Nutrient Overload Cascade
When caloric intake consistently exceeds energy demand, mitochondria face a bioenergetic bottleneck. Here is the chain of events:
Excess glucose and fatty acids flood the ETC
Membrane potential (Δψ) rises abnormally high
Electrons escape the chain, forming superoxide radicals (ROS)
ROS accumulate and damage mitochondrial DNA (mtDNA)
ETC protein complexes degrade
ATP production becomes increasingly inefficient
The body compensates by storing more fat — worsening the cycle
The Overload Paradox
This cascade explains a phenomenon clinicians frequently observe: patients with obesity often experience chronic fatigue and low energy — not because they lack fuel, but because their mitochondria can no longer convert that fuel into ATP efficiently.
More calories → More oxidative stress → Less usable energy
This is the overload paradox, and it is central to understanding why simply "eating less" addresses the symptom without treating the underlying cellular dysfunction.
The Collapse of PGC-1α Signaling
Compounding this damage, chronic inflammation generated by ROS suppresses PGC-1α — the very protein needed to build replacement mitochondria. This creates a self-reinforcing cycle from which the cell struggles to escape without deliberate intervention.
5. Tissue-by-Tissue Breakdown: Where Things Go Wrong
One of the most important insights from recent research is that mitochondrial dysfunction does not manifest uniformly. Different tissues experience distinct patterns of damage — and each contributes to obesity in its own way.
White Adipose Tissue: Where Fat Storage Goes Wrong
Fat cells (adipocytes) are metabolically active, hormone-secreting structures. Research published in Acta Biochimica et Biophysica Sinica (Xu et al., 2025) shows that mitochondrial dysfunction during adipocyte differentiation promotes:
Adipocyte hypertrophy — fat cells swell abnormally rather than multiplying
Local hypoxia — enlarged fat depots become oxygen-starved
Loss of "beige" adipocytes — a subtype of fat cell that burns calories as heat through a process called thermogenesis
The loss of beige fat is particularly significant: these specialized cells represent a meaningful calorie-burning mechanism, and obesity systematically dismantles the conditions needed to maintain them.
Not All Fat Is Created Equal
Visceral fat (around organs) shows more severe mitochondrial dysfunction than subcutaneous fat (under the skin), including greater reductions in oxidative capacity and higher ROS production — aligning with its stronger association with cardiometabolic risk (Das et al., 2024).
Skeletal Muscle: The Insulin Resistance Factory
Skeletal muscle is responsible for the majority of glucose uptake after a meal. When muscle mitochondria cannot fully oxidize fatty acids, the incomplete breakdown products — ceramides and diacylglycerols (DAGs) — accumulate inside muscle fibers.
These lipid intermediates are directly toxic to insulin signaling. They activate stress-sensitive kinases (PKCθ, JNK) that phosphorylate IRS-1 at inhibitory sites, blocking the GLUT4 translocation response to insulin. In plain terms:
The "door" that lets glucose into muscle cells gets jammed — and blood sugar stays elevated.
This is one of the most well-characterized cellular mechanisms linking mitochondrial dysfunction to type 2 diabetes.
The Liver: A Failing Adaptation
The liver initially compensates for increased fat influx by upregulating mitochondrial activity. But this adaptation is finite. As dysfunction progresses, the liver's ability to oxidize fat fails — driving the progression from simple fatty liver → NAFLD → NASH and increasing systemic lipid spillover.
The Brain: The Most Surprising Culprit
Perhaps the most paradigm-shifting finding of recent years involves the brain.
Research published in Biochimica et Biophysica Acta (Luo et al., 2025) demonstrated that POMC neurons — specialized hypothalamic neurons that form the brain's primary appetite-suppression circuit — are exquisitely sensitive to mitochondrial dysfunction.
When these neurons lose mitochondrial integrity, they fail to fire in response to satiety signals. The result:
Hunger persists even after adequate caloric intake
The feeling of fullness is blunted
Weight regain after dieting becomes biologically driven, not just psychological
"Mitochondrial dysfunction in POMC neurons contributes to the regulation of obesity, suggesting that the brain's energy-sensing machinery is itself a target of metabolic disease — not merely a bystander." — Luo et al., 2025
This finding fundamentally reframes the "willpower narrative" around obesity.
6. The Vicious Cycle: Inflammation, Damaged DNA & Cellular Chaos
Mitochondrial dysfunction does not stay local. It triggers a system-wide inflammatory cascade that makes metabolic recovery progressively harder.
When the Cleanup System Fails
Normally, the PINK1–Parkin mitophagy pathway identifies and recycles damaged mitochondria before they cause harm. In obesity, this quality-control system becomes overwhelmed — allowing defective mitochondria to accumulate and continue generating ROS.
Mitochondrial DNA as a Danger Signal
When damaged mitochondria rupture, they release mtDNA into the cytoplasm. Because mitochondria evolved from ancient bacteria, their DNA contains unmethylated CpG motifs similar to bacterial DNA.
The immune system recognizes this escaped mtDNA as a Damage-Associated Molecular Pattern (DAMP) — treating it like a bacterial invasion. This activates:
The NLRP3 inflammasome — a major driver of chronic low-grade inflammation
The cGAS-STING pathway — amplifying inflammatory cytokine production (IL-1β, TNF-α)
A comprehensive 2025 review in Biomolecules (Marino et al.) mapped this cascade from peripheral tissue mitochondrial impairment through to neuroinflammation in the central nervous system — demonstrating that the inflammatory consequences are not local but systemic.
The Self-Reinforcing Loop
Chronic inflammation from mtDNA release further suppresses PGC-1α, reduces mitophagy efficiency, and increases ROS production — completing a vicious cycle that becomes increasingly difficult to interrupt without deliberate intervention.
7. New Frontiers: 2025–2026 Research You Should Know
The science of mitochondrial dysfunction in obesity is evolving rapidly. Several 2025–2026 studies have opened important new directions:
Paternal Obesity and Intergenerational Mitochondrial Effects
A 2026 study published in Nature Communications (Huang, Park, Altıntaş et al.) made a striking discovery: male obesity can transmit mitochondrial dysfunction to offspring via a let-7–DICER molecular axis in adipose tissue. This finding suggests that mitochondrial health has intergenerational consequences that extend beyond the individual.
Endothelial NOX1: A Vascular Driver
Research in Circulation Research (Huang, Huang, Zhang et al., 2026) identified endothelial NOX1 — an enzyme in blood vessel walls — as a driver of obesity via skeletal muscle mitochondrial dysfunction. This highlights a vascular-metabolic connection that was previously underappreciated.
Drosophila Models Reveal Universal Mechanisms
A 2026 review in Frontiers in Endocrinology (Zeng et al.) using Drosophila models confirmed that altered mitochondrial functionality in metabolic disorders follows evolutionarily conserved pathways — strengthening confidence in the mechanisms identified in human studies.
Nutritional Epigenetics of Mitochondrial Function
A 2026 paper in Genes & Nutrition (Goss & Voruganti) explored how nutri-epigenetics — the way diet shapes gene expression — influences mitochondrial function and energy homeostasis in obesity, pointing toward precision nutrition as a future therapeutic avenue.
8. Seven Evidence-Based Strategies to Restore Mitochondrial Health
The most important clinical implication of this science: mitochondria are modifiable. Their dysfunction is not permanent. Here are the most powerful strategies supported by current evidence.
Strategy 1: Exercise — The Most Potent Mitochondrial Medicine
Physical activity is the single most effective intervention for mitochondrial health. Exercise:
Activates PGC-1α, triggering the production of new mitochondria (biogenesis)
Enhances mitophagy, clearing damaged organelles
Improves ETC efficiency and reduces ROS generation
Protocol:
150 minutes/week of moderate-intensity aerobic exercise (Zone 2 training — conversational pace)
2 sessions/week of resistance training
Zone 2 specifically targets fat oxidation and mitochondrial efficiency
"Exercise is the only intervention that forces your body to build brand-new, high-efficiency mitochondria — and upgrades the existing ones simultaneously."
Strategy 2: Time-Restricted Eating (TRE)
Giving your mitochondria a break from constant nutrient influx allows cellular repair systems to activate.
Protocol: Maintain a consistent 10–12 hour eating window (e.g., 8 AM – 6 PM or 7 AM – 7 PM)
Mechanism: Fasting activates AMPK and SIRT1/Sirtuins, which:
Stimulate mitophagy and mitochondrial biogenesis
Improve insulin sensitivity
Reduce oxidative stress
Note: Extended fasting protocols should be discussed with a healthcare provider, particularly for those with diabetes or other metabolic conditions.
Strategy 3: Anti-Inflammatory, Pro-Mitochondrial Diet
The building blocks of mitochondrial membranes come from your diet.
Focus on:
Omega-3 fatty acids (fatty fish, flaxseed, walnuts) — reduce membrane oxidation
Polyphenols (berries, green tea, extra-virgin olive oil, dark chocolate) — activate SIRT1 and reduce ROS
Magnesium (leafy greens, nuts, seeds) — essential cofactor for ATP synthesis
B vitamins (whole grains, legumes, eggs) — critical for ETC function
Limit:
Ultra-processed foods high in refined carbohydrates
Trans fats and excessive saturated fat
Alcohol (directly impairs mitochondrial function)
Strategy 4: Optimize Sleep
Deep sleep is when your body performs its most intensive mitochondrial repair.
Protocol: Prioritize 7–9 hours of quality sleep per night
Why it matters: Chronic sleep deprivation raises oxidative stress, suppresses mitophagy, and produces a mitochondrial profile similar to that seen with a high-fat diet. Even one week of sleep restriction measurably impairs metabolic flexibility.
Strategy 5: Thermal Stress (Sauna & Cold Exposure)
Controlled exposure to extreme temperatures acts as a hormetic stressor — mild cellular stress that triggers adaptive responses.
Heat (sauna, 15–20 min, 3–4x/week): Activates heat shock proteins that protect mitochondrial structure and folding
Cold exposure (cold showers, ice baths): Stimulates beige fat recruitment and activates uncoupling proteins that improve thermogenesis
Note: Thermal stress protocols should be approached cautiously by those with cardiovascular conditions. Consult your physician first.
Strategy 6: Targeted Supplementation
While lifestyle remains the foundation, specific nutraceuticals ("mitoceuticals") can support mitochondrial function as adjuncts:
Here is the supplement breakdown rewritten into a clean, scannable, and highly authoritative bulleted format.
CoQ10 (Ubiquinol)
Mechanism: Functions as an essential electron carrier within the mitochondrial electron transport chain (ETC).
Evidence Level: Moderate–Strong
NAD+ Precursors (NMN, NR)
Mechanism: Elevates intracellular NAD+ levels to support SIRT1 pathway activation and mitochondrial DNA (mtDNA) repair.
Evidence Level: Moderate
MitoQ
Mechanism: Acts as a specialized, mitochondria-targeted antioxidant designed to penetrate the inner mitochondrial membrane and reduce localized oxidative stress.
Evidence Level: Emerging
Alpha-Lipoic Acid (ALA)
Mechanism: Functions as a potent antioxidant that directly neutralizes free radicals and reduces oxidative stress within the ETC.
Evidence Level: Moderate
Magnesium Glycinate
Mechanism: Serves as a critical cofactor bound to ATP, rendering the synthesized energy biologically active and usable by the cell.
Evidence Level: Strong
Always consult a physician before starting supplements, particularly if you take medications for diabetes, hypertension, or heart disease.
Strategy 7: Stress Management
Chronic psychological stress elevates cortisol, which suppresses mitochondrial biogenesis and promotes visceral fat accumulation. Evidence supports:
Mindfulness-based stress reduction (MBSR)
Yoga and breathwork (activates parasympathetic pathways)
Nature exposure (shown to reduce inflammatory markers)
9. Key Biomarkers to Track Mitochondrial and Metabolic Health
You cannot optimize what you do not measure. These clinical markers provide a window into your mitochondrial health:
Fasting Insulin
What It Reflects: Baseline insulin sensitivity and metabolic flexibility.
Optimal Range: Less than 5 μIU/mL
Fasting Glucose
What It Reflects: Acute glucose metabolism and regulation.
Optimal Range: 70–99 mg/dL
HOMA-IR (Homeostatic Model Assessment for Insulin Resistance)
What It Reflects: Calculated index of underlying insulin resistance.
Optimal Range: Less than 1.5
Triglycerides
What It Reflects: Fat storage patterns and general lipid oxidation efficiency.
Optimal Range: Less than 100 mg/dL
Triglyceride-to-HDL Ratio
What It Reflects: A highly accurate lipid proxy for insulin resistance.
Optimal Range: Less than 2.0
Waist Circumference
What It Reflects: Visceral fat accumulation (a direct clinical proxy for mitochondrial fragmentation risk).
Optimal Range: Less than 94 cm for men, less than 80 cm for women
HbA1c (Hemoglobin A1c)
What It Reflects: Three-month weighted average of blood glucose concentrations.
Optimal Range: Less than 5.7%
hs-CRP (High-Sensitivity C-Reactive Protein)
What It Reflects: Systemic low-grade inflammation (often driven by mitochondrial DAMP leakage).
Optimal Range: Less than 1.0 mg/LDiscuss these tests with your healthcare provider in the context of your individual health history and risk factors.
10. Common Myths About Metabolism and Weight Loss
Myth 1: "Obesity is purely a willpower problem."
Reality: Mitochondrial dysfunction in POMC neurons biologically impairs satiety signaling. Hunger dysregulation in obesity has a measurable cellular basis, not just a behavioral one.
Myth 2: "Eating less is always enough to fix metabolism."
Reality: Calorie restriction without addressing mitochondrial dysfunction can further suppress PGC-1α and reduce metabolic rate. Restoring cellular energy production requires exercise, sleep, and dietary quality — not just quantity reduction.
Myth 3: "Supplements alone can fix mitochondrial dysfunction."
Reality: No supplement can replace the mitochondrial biogenesis stimulus that comes from regular physical activity. Supplements are adjuncts, not foundations.
Myth 4: "All body fat is equally harmful."
Reality: Visceral fat depots show markedly more severe mitochondrial dysfunction and inflammatory activity than subcutaneous fat — and carry significantly greater cardiometabolic risk.
Myth 5: "Metabolic damage is permanent."
Reality: Mitochondria exhibit remarkable plasticity. With consistent lifestyle intervention, mitochondrial density, function, and metabolic flexibility can be meaningfully restored — at virtually any age.
Myth 6: "Feeling tired means you need more calories."
Reality: Fatigue in obesity often reflects impaired ATP production from dysfunctional mitochondria — not caloric insufficiency. The solution is restoring mitochondrial efficiency, not increasing caloric intake.
11. Evidence Summary Table
Xia et al. (2024) | Nature Metabolism
Key Discovery: Identified that the signaling protein RalA drives destructive mitochondrial fragmentation in white adipose tissue by activating the fission protein DRP1.
Clinical Relevance: Establishes a direct molecular pathway explaining why fat cells lose their structure and energy capacity during chronic overnutrition.
Tung et al. (2024) | Current Obesity Reports
Key Discovery: Demonstrated that a lower mitochondrial DNA (mtDNA) copy number directly correlates with visceral adiposity and systemic insulin resistance.
Clinical Relevance: Highlights mtDNA depletion as a measurable biomarker for severe metabolic dysfunction and belly fat accumulation.
Das et al. (2024) | International Journal of Molecular Sciences
Key Discovery: Confirmed that visceral fat depots (deep belly fat) exhibit significantly greater mitochondrial decay and dysfunction than subcutaneous fat depots (fat under the skin).
Clinical Relevance: Proves that fat location dictates its toxicity; visceral fat actively destroys its own power plants, driving systemic disease.
Luo et al. (2025) | BBA – Molecular Basis of Disease
Key Discovery: Showed that localized mitochondrial failure within POMC neurons in the brain directly impairs central satiety signaling.
Clinical Relevance: Confirms that the physical inability to feel full is driven by cellular energy failures in the brain's appetite control centers.
Xu et al. (2025) | Acta Biochimica et Biophysica Sinica
Key Discovery: Discovered that underlying mitochondrial dysfunction completely disrupts healthy adipocyte maturation and differentiation.
Clinical Relevance: Explains how defective cells prevent the body from safely storing excess fat, forcing lipids to spill into toxic locations like the liver and muscles.
Marino et al. (2025) | Biomolecules
Key Discovery: Demonstrated a systemic link where peripheral mitochondrial dysfunction drives neuroinflammation throughout the central nervous system.
Clinical Relevance: Provides the explicit molecular connection between metabolic breakdown, cognitive decline, and chronic "brain fog."
Huang et al. (2026) | Nature Communications
Key Discovery: Proved that paternal obesity epigenetically transmits mitochondrial dysfunction to first-generation (F1) offspring via a specific let-7–DICER microRNA pathway.
Clinical Relevance: Establishes that a father's metabolic health before conception physically alters the cellular power plants of his children.
Huang et al. (2026) | Circulation Research
Key Discovery: Revealed that blood vessel inflammation (via endothelial NOX1) drives weight gain by poisoning mitochondrial function within nearby skeletal muscle cells.
Clinical Relevance: Connects cardiovascular health directly to metabolic rate, proving that damaged blood vessels ruin a muscle's ability to burn fat.
Goss & Voruganti (2026) | Genes & Nutrition
Key Discovery: Mapped how specific nutritional inputs directly rewrite mitochondrial gene expression through distinct nutritional-epigenetic pathways.
Clinical Relevance: Confirms that dietary choices act as direct molecular commands that can either repair or degrade cellular energy architecture over time.
12. Frequently Asked Questions
Q1: What is the main connection between mitochondrial dysfunction and obesity?
Chronic overnutrition overloads the mitochondrial electron transport chain, raising membrane potential beyond optimal levels. This causes electrons to leak and form reactive oxygen species (ROS), which damage mitochondrial DNA, impair ATP production, and reduce metabolic efficiency — creating a state of cellular energy insufficiency despite caloric excess.
Q2: Can mitochondrial dysfunction be reversed?
Yes. Mitochondria are highly plastic organelles. Regular aerobic exercise (particularly Zone 2 training) is the most potent trigger of mitochondrial biogenesis via PGC-1α activation. Combined with time-restricted eating, sleep optimization, and an anti-inflammatory diet, significant improvement in mitochondrial function is achievable.
Q3: How does mitochondrial dysfunction cause insulin resistance?
When muscle mitochondria cannot fully oxidize fatty acids, toxic lipid intermediates — ceramides and diacylglycerols (DAGs) — accumulate inside muscle fibers. These activate kinases (PKCθ, JNK) that block the insulin signaling cascade at IRS-1, preventing glucose uptake.
Q4: What are the signs of mitochondrial dysfunction in obesity?
Clinical indicators include persistent fatigue despite adequate sleep, difficulty losing weight despite calorie restriction, high fasting insulin, elevated triglycerides, low HDL cholesterol, central (visceral) adiposity, and poor exercise tolerance. These overlap significantly with metabolic syndrome.
Q5: Does exercise really improve mitochondrial function?
Strongly yes. Physical activity is the most potent biological trigger for PGC-1α — the master regulator of mitochondrial biogenesis. Exercise simultaneously increases mitochondrial density, improves respiratory chain efficiency, and enhances mitophagy (the recycling of damaged mitochondria).
Q6: Why do people with obesity feel tired if they have excess energy stored?
This reflects the "overload paradox." While fat stores are abundant, dysfunctional mitochondria cannot efficiently convert those stores into ATP. The result is subjective fatigue and low energy — despite, not because of, caloric surplus.
Q7: What role does the brain play in mitochondrial dysfunction and obesity?
POMC neurons in the hypothalamus — the brain's primary appetite-suppression circuit — are highly sensitive to mitochondrial dysfunction. When these neurons lose mitochondrial integrity, they fail to generate adequate satiety signals, biologically promoting continued food intake and weight gain.
Q8: Are there genetic factors in mitochondrial dysfunction?
Yes. Mitochondrial DNA is maternally inherited and subject to mutation from oxidative stress. Additionally, a 2026 study demonstrated that paternal obesity can transmit mitochondrial dysfunction to offspring via epigenetic mechanisms (the let-7–DICER axis) — suggesting intergenerational metabolic risk.
Q9: What supplements actually support mitochondrial health?
The strongest evidence exists for CoQ10 (ubiquinol form), NAD+ precursors (NMN or NR), and magnesium. MitoQ shows promise in emerging research. Supplements should be viewed as adjuncts to lifestyle intervention, not replacements. Consult a healthcare provider before starting any supplementation protocol.
Q10: How is visceral fat different from subcutaneous fat in terms of mitochondrial health?
Research shows visceral adipose tissue (fat around organs) exhibits significantly more severe mitochondrial dysfunction than subcutaneous fat — including greater reductions in oxidative capacity and higher ROS production. This aligns with visceral fat's stronger association with insulin resistance, cardiovascular disease, and type 2 diabetes.
Q11: What is metabolic flexibility, and why does it matter?
Metabolic flexibility is the ability to efficiently switch between burning carbohydrates and fat as fuel sources. Healthy mitochondria enable this flexibility; dysfunctional mitochondria lose it. Loss of metabolic flexibility is a hallmark of insulin resistance, obesity, and type 2 diabetes.
Q12: How does sleep deprivation affect mitochondrial function?
Chronic sleep deprivation raises oxidative stress, suppresses mitophagy, and impairs PGC-1α signaling — producing a mitochondrial profile similar to that seen with chronic high-fat feeding. Just one week of restricted sleep measurably reduces metabolic flexibility in otherwise healthy adults.
13. Editorial: Mitochondrial Dysfunction and the Biology of Obesity
Obesity is no longer a disease of excess calories alone—it is a disease of cellular energy failure. At the center of this paradigm shift lies the mitochondrion, whose dysfunction reframes obesity as a disorder of impaired energy processing rather than simple energy imbalance.
Chronic overnutrition creates a bioenergetic bottleneck. Excess substrates flood the electron transport chain, elevating membrane potential (Δψ) beyond physiological thresholds, leading to electron leak and overproduction of reactive oxygen species (ROS). This marks the transition from efficient ATP generation to oxidative stress–driven damage.
The “overload paradox” defines modern metabolic disease. More fuel does not translate into more usable energy; instead, it degrades mitochondrial efficiency, damages mtDNA, and impairs oxidative phosphorylation—creating a state of cellular energy insufficiency despite caloric excess.
Mitochondrial dysfunction is tissue-specific and systemically integrated. In adipose tissue, it drives hypertrophy and inflammation; in skeletal muscle, it promotes lipid-induced insulin resistance; in the liver, it accelerates steatosis; and in the hypothalamus—particularly within POMC neurons—it disrupts satiety signaling, biologically reinforcing weight gain.
Failure of mitochondrial quality control amplifies disease progression. Impaired mitophagy (via PINK1–Parkin pathways) leads to accumulation of damaged mitochondria, while released mitochondrial DNA activates the NLRP3 inflammasome, linking bioenergetic failure to chronic low-grade inflammation.
At the molecular level, suppression of PGC-1α represents a critical inflection point. Reduced mitochondrial biogenesis prevents recovery, locking cells into a state of declining metabolic flexibility.
Therapeutically, mitochondria are not passive victims—they are modifiable targets. Exercise remains the most potent activator of mitochondrial biogenesis and oxidative capacity, while fasting-related pathways (AMPK, SIRT1) restore quality control mechanisms.
The clinical implication is profound: obesity must be approached as a systems-level mitochondrial disease. Effective management requires restoring energy flux, not merely restricting caloric intake.
Future medicine will likely target mitochondrial networks directly, integrating pharmacology, lifestyle, and precision biomarkers to reverse metabolic dysfunction at its cellular roots.
Your Evidence-Based Action Plan
Priority 1 — Start Moving (This Week) Commit to 150 minutes of moderate aerobic exercise per week. Zone 2 training (conversational intensity) specifically targets mitochondrial efficiency and fat oxidation.
Priority 2 — Establish an Eating Window (This Month) Adopt a 10–12 hour eating window consistently. This simple practice activates AMPK and Sirtuin pathways that restore mitochondrial quality control.
Priority 3 — Audit Your Diet (Ongoing) Increase omega-3s, polyphenols, magnesium, and B vitamins. Reduce ultra-processed foods, refined carbohydrates, and alcohol.
Priority 4 — Protect Your Sleep (Always) Treat 7–9 hours of quality sleep as non-negotiable. It is when your cells repair what the day has damaged.
Priority 5 — Measure What Matters (Every 3–6 Months) Work with your healthcare provider to track fasting insulin, HOMA-IR, triglycerides, waist circumference, and hsCRP. These numbers tell the story of your mitochondrial health.
Priority 6 — Consider Thermal Stress (When Ready). Once foundational habits are established, explore sauna and/or cold exposure as additional mitochondrial stressors — under medical guidance if you have cardiovascular concerns.
Priority 7 — Discuss Targeted Supplementation (With Your Doctor) If lifestyle measures alone are insufficient, discuss CoQ10, NAD+ precursors, or other mitoceuticals with a qualified healthcare provider.
Author’s Note (Clinician’s Perspective)
As a clinician in internal medicine, one of the most important shifts I have witnessed over the past decade is the transition from viewing obesity as a lifestyle issue to recognizing it as a complex, systems-level metabolic disease rooted in cellular dysfunction. Among the many mechanisms involved, mitochondrial health has emerged as a central and unifying theme.
In daily clinical practice, patients rarely present with “mitochondrial dysfunction” as a diagnosis. Instead, they come with type 2 diabetes, fatty liver disease, central obesity, dyslipidemia, hypertension, or unexplained fatigue. Yet, when we look deeper, these seemingly different conditions often share a common biological thread: impaired mitochondrial energy production, increased oxidative stress, chronic inflammation, and reduced metabolic flexibility.
I frequently encounter patients who tell me, “Doctor, I eat less than before, but I still gain weight and feel exhausted.” One patient in his mid-50s presented with abdominal obesity, elevated triglycerides, prediabetes, and persistent fatigue despite multiple attempts at dieting. He believed his lack of success reflected poor willpower. However, laboratory findings revealed severe insulin resistance and metabolic dysfunction. After implementing a structured program that emphasized aerobic and resistance exercise, improved sleep, nutritional quality, and gradual weight reduction, his insulin sensitivity improved, energy levels increased, and several metabolic markers normalized. His experience illustrates an important reality: obesity is often driven by underlying biological dysfunction rather than a lack of motivation.
What is particularly important—and often misunderstood—is that these mitochondrial changes are not merely consequences of obesity; they are active drivers of disease progression. Dysfunctional mitochondria in skeletal muscle contribute to insulin resistance. In adipose tissue, they promote inflammation and unhealthy fat expansion. In the liver, they accelerate metabolic dysfunction and fatty liver disease. In the brain, particularly within POMC neurons, they alter appetite regulation in ways that make sustained weight loss biologically challenging.
From a treatment standpoint, this perspective changes everything. It shifts our focus away from short-term calorie restriction toward long-term restoration of metabolic health. Interventions such as structured exercise, sleep optimization, stress reduction, and targeted nutritional strategies are not simply “lifestyle advice”—they are mitochondrial therapies with measurable biochemical effects. Even many modern pharmacological treatments exert part of their benefit through pathways that influence mitochondrial function and cellular energy metabolism.
Equally important is patient communication. When individuals understand that their condition is rooted in complex biological adaptations rather than personal failure, feelings of guilt and stigma often diminish. This understanding frequently improves engagement, adherence, and long-term success.
Ultimately, addressing obesity requires more than managing body weight—it requires restoring the body's ability to efficiently produce, utilize, and regulate energy at the cellular level. That is where meaningful and lasting clinical outcomes begin. Through evidence-based exercise, sound nutrition, adequate sleep, stress management, appropriate medical therapy, and careful monitoring of key metabolic biomarkers, we can help patients improve not only their weight but also their overall metabolic health, quality of life, and long-term disease risk.
References
Das, S., Mukhuty, A., Mullen, G. P., & Rudolph, M. C. (2024). Adipocyte mitochondria: Deciphering energetic functions across fat depots in obesity and type 2 diabetes. International Journal of Molecular Sciences, 25(12), Article 6681. https://doi.org/10.3390/ijms25126681
Goss, L. R., & Voruganti, V. S. (2026). Nutri-epigenetics of mitochondrial function and energy homeostasis in obesity. Genes & Nutrition, 21, Article 1. https://doi.org/10.1186/s12263-025-00781-w
Huang, C., Park, J. H., Altıntaş, A., et al. (2026). Male obesity causes adipose mitochondrial dysfunction in F1 mouse progeny via a let-7–DICER axis. Nature Communications, 17, Article 3125. https://doi.org/10.1038/s41467-026-69686-5
Huang, K., Huang, Y., Zhang, Y., Hatch, N. W., Freed, J. K., & Cai, H. (2026). Endothelial NOX1 drives obesity via skeletal muscle mitochondrial dysfunction. Circulation Research. Advance online publication. https://doi.org/10.1161/CIRCRESAHA.125.326768
Luo, X.-d., Tang, S., Luo, X.-y., Luosang, Q., Xia, R.-h., & Wang, X.-w. (2025). Mitochondrial regulation of obesity by POMC neurons. Biochimica et Biophysica Acta (BBA) — Molecular Basis of Disease, 1871(3), Article 167682. https://doi.org/10.1016/j.bbadis.2025.167682
Marino, F., Petrella, L., Cimmino, F., Pizzella, A., Monda, A., Allocca, S., Rotondo, R., D'Angelo, M., Musco, N., Iommelli, P., Catapano, A., Bagnato, C., Paolini, B., & Cavaliere, G. (2025). From obesity to mitochondrial dysfunction in peripheral tissues and in the central nervous system. Biomolecules, 15(5), Article 638. https://doi.org/10.3390/biom15050638
Tung, P. W., Thaker, V. V., Gallagher, D., et al. (2024). Mitochondrial health markers and obesity-related health in human population studies: A narrative review of recent literature. Current Obesity Reports, 13, 724–738. https://doi.org/10.1007/s13679-024-00588-7
Xia, W., Veeragandham, P., Cao, Y., Xu, Y., Rhyne, T. E., Qian, J., Hung, C. W., Zhao, P., Jones, Y., Gao, H., Liddle, C., Yu, R. T., Downes, M., Evans, R. M., Rydén, M., Wabitsch, M., Wang, Z., Hakozaki, H., Schöneberg, J., Reilly, S. M., … Saltiel, A. R. (2024). Obesity causes mitochondrial fragmentation and dysfunction in white adipocytes due to RalA activation. Nature Metabolism, 6(2), 273–289. https://doi.org/10.1038/s42255-024-00978-0
Xu, Y., Xie, S., Han, L., Xu, L., & Zhu, Y. (2025). Mitochondrial dysfunction in adipocyte differentiation: Implications for obesity and metabolic syndrome intervention. Acta Biochimica et Biophysica Sinica. Advance online publication. https://doi.org/10.3724/abbs.2025153
Zeng, Y., Yang, Y., Zhu, Y., Xu, P., Cheong, C., Zhao, Y. J., & Baeg, G. H. (2026). Altered mitochondrial functionality in metabolic disorders: Insights from Drosophila studies. Frontiers in Endocrinology, 17, Article 1817121. https://doi.org/10.3389/fendo.2026.1817121
.Last reviewed: June 2026. This article will be updated as new research emerges.
Author: Dr. T.S. Didwal, M.D. (Internal Medicine)