Fuel Partitioning Explained: How Your Body Decides to Burn Fat or Sugar
Why does your body burn sugar instead of fat? Learn how fuel partitioning and insulin signaling shape metabolic health and disease
METABOLISM
Dr. T.S. Didwal, M.D.(Internal Medicine)
3/1/202613 min read
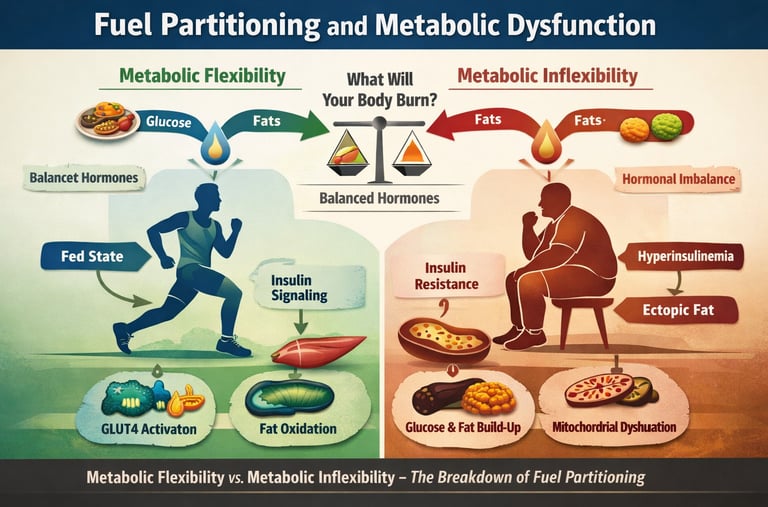
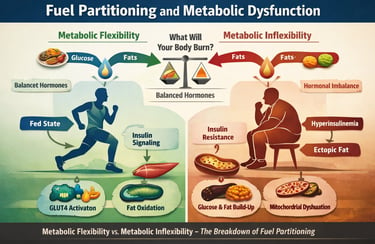
What if the real driver of cardiometabolic disease is not high blood glucose, but a silent failure in fuel partitioning?
At every moment, your cells are making a metabolic decision: burn glucose, oxidise fatty acids, or conserve energy for later use. This decision is governed by a tightly regulated hormonal network involving insulin signaling, counterregulatory hormones, mitochondrial substrate competition, and the dynamic process known as metabolic flexibility. In healthy physiology, this system functions with remarkable precision. After a meal, insulin activates the PI3K–AKT pathway, promotes GLUT4 translocation, stimulates glycogenesis and lipogenesis, and suppresses lipolysis — directing energy into storage while preventing glucotoxicity (Burchfield et al., 2025). During fasting or exercise, glucagon, catecholamines, and cortisol reverse this program, restoring fatty acid oxidation and hepatic glucose output (Huang et al., 2025).
But when chronic caloric excess, sedentary behavior, or visceral adiposity disrupt this orchestration, the system begins to misfire. Skeletal muscle — responsible for the majority of insulin-stimulated glucose disposal — becomes resistant to insulin. Mitochondrial oxidative capacity declines. Fatty acids accumulate ectopically. The biochemical competition between glucose and lipid oxidation, classically described by the Randle cycle, becomes pathologically skewed (Challa, 2025). What emerges is metabolic inflexibility — the inability to appropriately switch between substrates — now recognized as an early hallmark of insulin resistance, hyperinsulinemia, and cardiometabolic disease (Ang et al., 2025).
Crucially, hyperinsulinemia itself may drive vascular dysfunction independent of glucose levels, challenging the long-standing glucocentric model of type 2 diabetes management (Fazio et al., 2024).
Understanding how the body decides what to burn — and what goes wrong — is therefore not academic. It is the key to preventing the metabolic collapse that underlies modern chronic disease.
Clinical pearls
1. Selective Insulin Resistance: The "Double-Edged" Signal
Scientific Brief: Insulin resistance is pathway-specific, not global. While the PI3K-AKT pathway (responsible for glucose uptake) becomes blunted, the MAPK/ERK pathway (responsible for cell growth and proliferation) often remains sensitive. This creates a state where high insulin fails to lower blood sugar but continues to drive vascular smooth muscle growth and atherosclerosis.
Think of insulin as a key that opens two different doors. In your body, the "sugar door" has a rusty lock and won't open, but the "growth door" is wide open. When your body pumps out extra insulin to force the sugar door, it accidentally over-activates the growth door, which can thicken your artery walls and increase heart disease risk.
2. The "Metabolic Lock-In" of Hyperinsulinemia
Scientific Brief: Chronically elevated insulin (hyperinsulinemia) acts as a potent inhibitor of adipose tissue lipolysis via the suppression of hormone-sensitive lipase. This creates a "metabolic lock" where the body cannot transition to fat oxidation (the Randle Cycle) even during caloric deficit, leading to perceived lethargy and "glucose dependency."
High insulin levels act like a "storage fat" padlock. Even if you eat less, if your insulin stays high, your body can't "unlock" your fat stores for fuel. This is why you might feel tired or hungry shortly after eating; your body is struggling to switch from burning sugar to burning its own stored fat.
3. Muscle as the "Metabolic Engine" of Flexibility
Scientific Brief: Skeletal muscle is the primary site of metabolic inflexibility. Early-stage dysfunction is characterized by reduced GLUT4 translocation and impaired mitochondrial switching. Interventions like Zone 2 endurance training specifically upregulate mitochondrial density, bypassing certain insulin-signaling defects through AMPK-mediated pathways.
Your muscles are like a hybrid car's engine. When you are healthy, you switch easily between gas (sugar) and electricity (fat). In early diabetes, that "switch" gets stuck. Regular, easy exercise (like a brisk walk) helps "re-train" your muscle mitochondria to switch fuels more efficiently, regardless of how much insulin you have.
4. Adiposity and the "Blunted" Glucagon Response
Scientific Brief: As shown by Huang et al. (2025), obesity significantly alters the counterregulatory axis. Obese T2DM patients often exhibit a blunted glucagon response to hypoglycemia but elevated basal cortisol. This "hormonal noise" makes blood sugar management more volatile and increases the risk of "brittle" glucose swings during treatment.
Carrying extra weight doesn't just change your looks; it changes how your "emergency" hormones work. In obesity, the hormone that prevents your blood sugar from dropping too low (glucagon) gets quiet, while your stress hormone (cortisol) stays loud. This makes it harder to keep your blood sugar in a steady, safe range.
5. Beyond the A1c: The Importance of Fasting Insulin
Scientific Brief: A normal HbA1c or fasting glucose can mask significant metabolic pathology. Hyperinsulinemia can precede hyperglycemia by a decade. Measuring fasting insulin and calculating HOMA-IR (Homeostatic Model Assessment for Insulin Resistance) provides a more proactive snapshot of cardiometabolic risk than glucose-centric metrics alone.
Looking only at your blood sugar (A1c) is like checking if your house is on fire. Checking your insulin levels is like checking if the wiring is smoking. Your blood sugar might look "normal" only because your body is working 10 times harder than it should to keep it there. Catching high insulin early lets us fix the problem before the "fire" of diabetes starts.
Fuel Partitioning & Hormonal Regulation: The Science of Metabolic Flexibility
How does the body decide whether to burn glucose or fat? This process, known as fuel partitioning, is a complex hormonal symphony. When in tune, the body shifts seamlessly between states. When disrupted—by hyperinsulinemia, insulin resistance, or sedentary living—the result is chronic cardiometabolic disease.
Drawing on landmark 2024–2025 research, this guide explores the molecular mechanisms of insulin signaling, the enduring relevance of the Randle Cycle, and why metabolic inflexibility is the earliest warning sign of systemic failure.
I. Insulin Signaling: Beyond a Linear Cascade
The traditional view of insulin as a simple "on/off" switch for blood sugar is obsolete. According to Burchfield et al. (2025) in Nature Metabolism, insulin operates via a "signaling network" rather than a linear chain.
The Receptor Tyrosine Kinase Pathway
Binding: Insulin binds to its receptor, triggering autophosphorylation.
Recruitment: Insulin Receptor Substrates (IRS) activate PI3K.
Activation: The second messenger PIP3 recruits AKT (Protein Kinase B), the central node of metabolic action.
GLUT4 Translocation and Glucose Clearance
The activation of AKT leads to the phosphorylation of AS160, releasing the "brake" on GLUT4 vesicles. These transporters move to the plasma membrane, increasing glucose uptake capacity by 10x. Because skeletal muscle handles 70–80% of this disposal, it remains the primary target for treating metabolic dysfunction.
II. Counterregulatory Hormones: The Fuel Reclaimers
While insulin directs storage, a group of "counterregulatory" hormones ensures fuel availability during fasting or stress.
Glucagon: The primary antagonist to insulin. It triggers hepatic glycogenolysis and gluconeogenesis. Recent findings by Huang et al. (2025) show that obese T2DM patients suffer from "blunted" glucagon responses, complicating glucose stability.
Catecholamines (Epinephrine/Norepinephrine): These drive rapid lipolysis and glycogenolysis during exercise while suppressing insulin secretion to protect brain glucose levels.
Cortisol: A long-term stress hormone that induces insulin resistance to prioritize cerebral glucose supply. High basal cortisol is now linked to worsened hepatic insulin resistance in obese populations (Huang et al., 2025).
III. The Randle Cycle and Metabolic Flexibility
The Randle Cycle (or glucose-fatty acid cycle) describes the competition between fuels. Challa (2025) argues in Nature Reviews Endocrinology that this 1960s framework is more relevant today than ever.
How the Body Switches Fuels
The Fed State: High insulin suppresses fat oxidation, forcing the body to burn glucose.
The Fasted State: Low insulin allows fatty acid oxidation. High-fat oxidation produces citrate, which inhibits glucose breakdown.
Metabolic Flexibility is the ability to switch between these states efficiently. Metabolic Inflexibility—the failure to suppress fat burning after a meal—is the hallmark of early insulin resistance (Ang et al., 2025).
IV. Pathological Disruptions: Hyperinsulinemia & Resistance
When fuel partitioning fails, two distinct but related pathologies emerge:
1. The Danger of Hyperinsulinemia
Fazio et al. (2024) argue that chronically high insulin is an independent risk factor for heart disease. Even if blood sugar is "normal," hyperinsulinemia drives:
Vascular smooth muscle proliferation.
Endothelial dysfunction.
Sympathetic nervous system overactivation.
2. Selective Insulin Resistance
Insulin resistance isn't always "all or nothing." In Selective Insulin Resistance, the pathway for glucose disposal (PI3K-AKT) is blocked, but the pathway for cell growth (MAPK/ERK) remains active. This explains why patients can have high blood sugar and accelerated arterial plaque simultaneously (Burchfield et al., 2025).
V. Clinical Insights: Restoring the "Metabolic Switch"
The transition from biochemistry to the bedside requires a focus on early biomarkers and tissue-specific interventions.
To restore the "metabolic switch" and improve fuel partitioning, clinical interventions must target specific tissues and molecular pathways. Here is a breakdown of the primary strategies based on the latest 2024–2025 research:
Endurance Exercise (Zone 2 Training)
Mechanism: Stimulates mitochondrial biogenesis and significantly upregulates GLUT4 expression.
Target Tissue: Primarily Skeletal Muscle, the body's largest glucose sink.
Result: Restores metabolic flexibility by bypassing certain insulin-signaling defects through AMPK-activated pathways.
Caloric Restriction and Weight Management
Mechanism: Reduces "lipotoxicity" by clearing diacylglycerols and ceramides—the toxic lipid metabolites that "clog" insulin signaling.
Target Tissue: Liver and Skeletal Muscle.
Result: Reverses the serine phosphorylation of IRS-1, allowing the insulin receptor to communicate effectively with the rest of the cell again.
GLP-1 Receptor Agonists (e.g., Semaglutide, Tirzepatide)
Mechanism: Enhances glucose-dependent insulin secretion while simultaneously improving satiety and reducing the total insulin burden over time.
Target Tissue: Systemic / Multi-organ (Brain, Pancreas, Gut, and Adipose).
Result: Lowers hyperinsulinemia and facilitates a transition out of "glucose-only" burning mode.
Low-Carbohydrate & Mediterranean Dietary Patterns
Mechanism: Minimizes postprandial insulin spikes, thereby reducing the chronic suppression of fat oxidation (the Randle Cycle).
Target Tissue: Adipose Tissue and the Vascular System.
Result: Protects the endothelium from the pro-inflammatory effects of high insulin and allows for easier access to stored body fat between meals.
Resistance Training (Hypertrophy)
Mechanism: Increases the total surface area and volume of tissue capable of insulin-mediated glucose disposal.
Target Tissue: Skeletal Muscle.
Result: Provides a larger "buffer" for glucose storage, reducing the need for the pancreas to overproduce insulin to maintain euglycemia.
Metabolic health requires looking beyond HbA1c. Measuring fasting insulin (HOMA-IR) and assessing mitochondrial capacity enable the detection of metabolic inflexibility decades before a T2DM diagnosis.
The 2026 Comprehensive Metabolic Health Panel
Fasting Insulin
Ideal Range: < 6 IU/mL
Why It Matters: This is the earliest indicator of hyperinsulinemia. Elevated levels signal a "locked" fat-burning switch, meaning your body is struggling to access stored energy.
HOMA-IR (Homeostatic Model Assessment for Insulin Resistance)
Ideal Range: < 1.0
Calculation: (Fasting Glucose × Fasting Insulin) / 405
Why It Matters: This ratio quantifies systemic insulin resistance. It tells us how much "effort" your pancreas must exert to keep your blood sugar stable.
TG/HDL Ratio
Ideal Range: < 1.5
Why It Matters: A powerful proxy for LDL particle size and a strong predictor of metabolic flexibility. A high ratio (over 2.5) suggests your cells are failing to burn fat efficiently.
ApoB (Apolipoprotein B)
Ideal Range: < 80 mg/dL
Why It Matters: While the article focuses on fuel, ApoB measures the actual number of "cargo ships" that cause arterial plaque. High insulin drives the production of these atherogenic particles.
Fasting Glucose
Ideal Range: 70–90 mg/dL
Why It Matters: While less sensitive than insulin, it remains the baseline for checking hepatic glucose output and ensuring the Randle Cycle is functioning.
HbA1c (Glycated Haemoglobin)
Ideal Range: 4.8\% – 5.4\%
Why It Matters: A 90-day average. In 2026, we interpret this alongside insulin to ensure your sugar is low because of efficiency, not because of high-pressure insulin compensation.
hs-CRP (High-Sensitivity C-Reactive Protein)
Ideal Range: < 1.0 mg/L
Why It Matters: Measures systemic low-grade inflammation. Inflammation is a primary "clog" that breaks the insulin receptor signaling network.
Uric Acid
Ideal Range: 3.0 – 5.5 mg/dL
Why It Matters: High levels can inhibit mitochondrial function. It is often a byproduct of high fructose intake or excessive insulin signaling, acting as a metabolic "canary in the coal mine."
Understanding the Results: Two Scenarios
The "Smoking Wire" (Compensated Resistance):
If your glucose is 85 mg/dL (normal) but your fasting insulin is 15 µIU/mL — Elevated (High Fasting Insulin)
Your body is working "overtime." This is a red flag that requires intervention years before the HbA1c would ever rise.
The "Metabolic Engine" (Flexible):
If your glucose is 85 mg/dL and your Fasting insulin is 4 µIU/mL (metabolically favourable level)
Your body is highly efficient. Your "metabolic switch" is working perfectly, allowing you to move between burning sugar and fat with ease.
Clinical Application
This panel allows for a stratified risk approach. A patient with a normal HbA1c but a HOMA-IR of 2.5 should be prioritised for the interventions —specifically Zone 2 training to boost mitochondrial density and low-carb protocols to lower the insulin burden.
Study Summaries and Key Takeaways
Insulin Resistance Is Pathway-Specific, Not Global
Burchfield, Diaz-Vegas, and James (2025) demonstrate that insulin signalling functions as a complex, multi-nodal network rather than a simple linear cascade. Feedback loops and cross-talk with mTORC1 and MAPK/ERK pathways explain why selective insulin resistance occurs — metabolic signaling (glucose disposal) becomes impaired, while mitogenic signaling remains active. This divergence helps explain the coexistence of metabolic dysfunction and accelerated cardiovascular disease.
Obesity Reshapes the Counterregulatory Hormone Environment
Huang et al. (2025) show that obese and non-obese men with type 2 diabetes exhibit markedly different hormonal profiles. Obese patients demonstrate blunted glucagon responses, elevated basal cortisol, and heightened sympathetic activity, a combination that worsens insulin resistance and increases cardiovascular risk. These findings support the need for adiposity-stratified treatment approaches.
Hyperinsulinemia Is an Independent Pathological Driver
Fazio et al. (2024) argue that chronic hyperinsulinemia independently promotes vascular disease, hypertension, and cancer risk, through pro-inflammatory, mitogenic, and sympathoexcitatory mechanisms — even when glucose levels are controlled. This challenges the traditional glucocentric model of diabetes management.
Metabolic Inflexibility Is the Earliest Detectable Defect
Ang et al. (2025) identify skeletal muscle metabolic inflexibility as an early event in cardiometabolic disease progression. Impaired mitochondrial oxidative capacity and reduced GLUT4 function precede overt hyperglycemia, suggesting that restoring fuel-switching capacity may offer the greatest preventive benefit.
Frequently Asked Questions (FAQs)
1. What is fuel partitioning, and why does it matter? Fuel partitioning refers to the processes by which the body decides which energy substrates — glucose, fatty acids, amino acids, or ketones — to oxidize or store in response to nutritional and hormonal signals. It matters because dysregulated partitioning underpins insulin resistance, obesity, type 2 diabetes, and cardiovascular disease. Understanding it allows clinicians and researchers to identify where the process breaks down and design targeted interventions.
2. How does insulin promote fat storage while also lowering blood sugar? These two effects arise from the same signaling cascade. Insulin activates AKT, which simultaneously promotes GLUT4 translocation (enabling glucose uptake), activates glycogen synthase (storing glucose as glycogen), and upregulates SREBP-1c (driving lipogenesis). Insulin also suppresses hormone-sensitive lipase in adipose tissue, halting fat breakdown. The net effect is that all ingested energy is directed toward storage rather than oxidation — a physiologically appropriate response to a meal that becomes problematic when chronically activated.
3. What distinguishes metabolic flexibility from metabolic inflexibility? Metabolic flexibility describes the capacity to switch efficiently between glucose and fat oxidation depending on substrate availability and hormonal context. A metabolically flexible person burns predominantly glucose after meals and shifts to fat during fasting or exercise. Metabolic inflexibility, as described by Ang et al. (2025), is the inability to make this switch efficiently — particularly the failure to suppress fat oxidation in the fed state or upregulate it in the fasted state — and is an early marker of insulin resistance and cardiometabolic risk.
4. Is the Randle cycle still scientifically relevant in 2025? Yes. As Challa (2025) argues in Nature Reviews Endocrinology, the Randle cycle's core insight — that fatty acid and glucose oxidation compete at multiple biochemical checkpoints — remains mechanistically accurate and clinically useful. It integrates observations from mitochondrial biology, lipid signaling, and whole-body metabolic phenotyping in a way that purely molecular frameworks cannot. Far from being obsolete, it serves as an indispensable organizing principle for understanding substrate competition in metabolic disease.
5. Can hyperinsulinemia cause cardiovascular disease even without diabetes? Evidence suggests yes. Fazio et al. (2024) compile data showing that chronically elevated insulin promotes vascular smooth muscle cell proliferation, endothelial dysfunction, sympathetic nervous system overactivation, and pro-inflammatory signaling — all independent risk factors for hypertension and atherosclerosis. Individuals with hyperinsulinemia and normal blood glucose can still carry substantial, underappreciated cardiovascular risk, which is why measuring fasting insulin and HOMA-IR may be more informative than glucose testing alone in certain populations.
6. Why do obese and non-obese type 2 diabetics have different counterregulatory hormone profiles? Adipose tissue is not metabolically inert — it secretes adipokines, contributes to systemic inflammation, and affects the hypothalamic-pituitary-adrenal (HPA) axis. Huang et al. (2025) found that obese T2DM patients have elevated basal cortisol and blunted glucagon responses compared to non-obese patients. Excess adiposity drives chronic low-grade inflammation that activates the HPA axis (elevating cortisol) while simultaneously disrupting α-cell glucagon secretion. These hormonal differences have direct implications for glycemic management, hypoglycemia risk, and cardiovascular outcome stratification.
7. What practical interventions best restore insulin sensitivity and metabolic flexibility? The most evidence-based interventions remain lifestyle-based: sustained aerobic exercise increases skeletal muscle mitochondrial biogenesis and GLUT4 expression; resistance training improves insulin-mediated glucose uptake; and caloric restriction reduces ectopic lipid accumulation. Dietary composition also matters — reducing ultra-processed carbohydrates lowers postprandial insulin demand. Pharmacologically, GLP-1 receptor agonists and SGLT-2 inhibitors show metabolic benefits that extend beyond glucose lowering, partly through mechanisms that reduce hyperinsulinemia and improve fat oxidation capacity.
Author’s Note
This article was written from a systems-level perspective on metabolic health. In clinical practice, we are often trained to view glucose as the central biomarker of metabolic disease. Yet over the past decade — and increasingly in the most recent literature — it has become clear that glucose is only the visible surface of a far deeper regulatory network.
Fuel partitioning, insulin signaling dynamics, counterregulatory hormone balance, mitochondrial oxidative capacity, and tissue-specific metabolic flexibility form an integrated physiological architecture. When that architecture is intact, metabolic health appears effortless. When it fragments — often silently, and years before diagnostic thresholds are crossed — disease emerges.
My intention in this piece was not merely to review recent studies, but to synthesize them into a coherent framework that connects molecular signaling with bedside reality. As a physician working at the intersection of metabolism, cardiovascular risk, and chronic disease, I believe we must move beyond a glucose-centric model toward a systems-based understanding of metabolic regulation. Hyperinsulinemia, selective insulin resistance, and early metabolic inflexibility deserve greater attention in both research and clinical risk assessment.
The concepts discussed here are grounded in peer-reviewed scientific literature from 2024–2025 and are intended for educational purposes. They are not a substitute for individualized medical evaluation.
Ultimately, the goal is simple: to reframe metabolic disease not as an abrupt failure of blood sugar control, but as a progressive loss of fuel adaptability — one that is measurable, understandable, and, in many cases, reversible.
Disclaimer: This article is for informational purposes only and does not constitute medical advice. Individual circumstances vary, and treatment decisions should always be made in consultation with qualified healthcare professionals.
Related Articles
Skeletal Muscle: The Missing Link in Blood Sugar Control and Metabolic Health | DR T S DIDWAL
Tired All Day, Awake at Night? The Redox and Mitochondrial Link | DR T S DIDWAL
Obesity and Fatty Liver Disease: What Science Says About Risk and Health | DR T S DIDWAL
Intermittent Fasting: Metabolic Health Benefits and the Evidence on Longevity | DR T S DIDWAL
Activate Your Brown Fat: A New Pathway to Longevity and Metabolic Health | DR T S DIDWAL
Leptin vs. Adiponectin: How Your Fat Hormones Control Weight and Metabolic Health | DR T S DIDWAL
References
Ang, J. C., Sun, L., Foo, S. R., Leow, M. K., Vidal-Puig, A., Fontana, L., & Dalakoti, M. (2025). Perspectives on whole body and tissue-specific metabolic flexibility and implications in cardiometabolic diseases. Cell Reports Medicine, 6(9), 102354. https://doi.org/10.1016/j.xcrm.2025.102354
Burchfield, J. G., Diaz-Vegas, A., & James, D. E. (2025). The insulin signalling network. Nature Metabolism, 7, 1745–1764. https://doi.org/10.1038/s42255-025-01349-z
Challa, A. A. (2025). The enduring relevance of the Randle cycle. Nature Reviews Endocrinology, 21, 269. https://doi.org/10.1038/s41574-025-01092-1
Fazio, S., Affuso, F., Cesaro, A., Tibullo, L., Fazio, V., & Calabrò, P. (2024). Insulin resistance/hyperinsulinemia as an independent risk factor that has been overlooked for too long. Biomedicines, 12(7), 1417. https://doi.org/10.3390/biomedicines12071417
Huang, R., Bao, Y., Xiong, Y., et al. (2025). Differences in the insulin counterregulatory hormones between obese and nonobese male patients with type 2 diabetes mellitus. Scientific Reports, 15, 12099. https://doi.org/10.1038/s41598-025-89543-7