De Novo Lipogenesis Explained: How Sugar Turns Into Liver Fat and High Triglycerides
Can sugar become saturated fat? Discover the science of de novo lipogenesis and how excess carbohydrates may fuel fatty liver, high triglycerides, and heart risk.
METABOLISM
Dr. T.S. Didwal, M.D.(Internal Medicine)
5/22/202624 min read
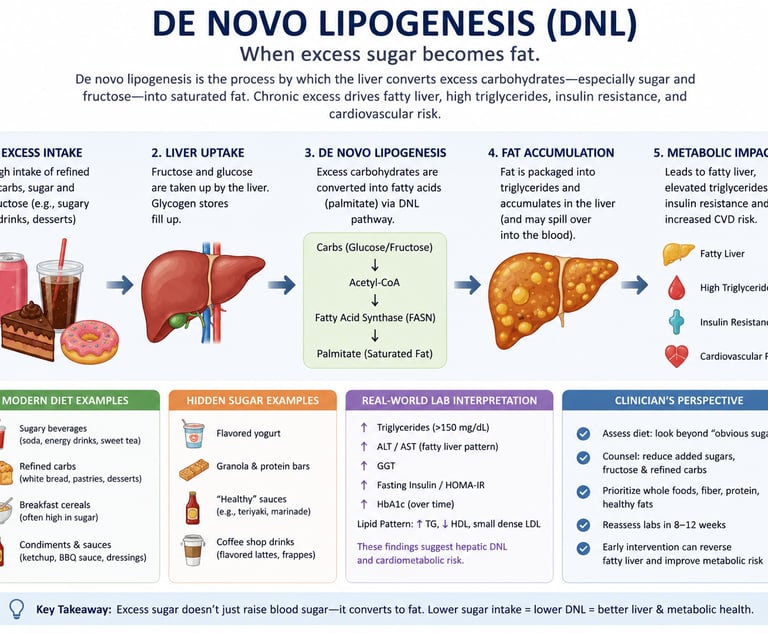
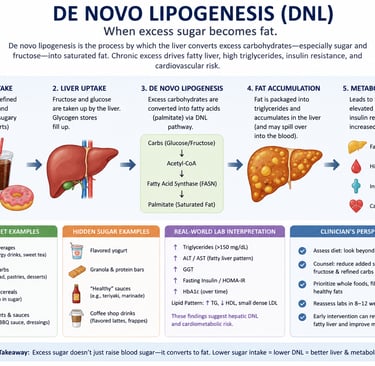
De novo lipogenesis (DNL) is the process by which the liver converts excess carbohydrates — especially sugar and fructose — into saturated fat. When glycogen stores are full, surplus glucose and fructose are transformed into fatty acids such as palmitate, which can contribute to fatty liver disease, elevated triglycerides, insulin resistance, and cardiovascular risk. High intake of refined carbohydrates and sugar-sweetened beverages is one of the strongest dietary drivers of hepatic DNL
Clinical Pearls
1. Your blood saturated fat levels often come from sugar, not from the fat you eat. High levels of palmitic acid (a saturated fat) in your bloodstream are frequently produced by your liver from excess sugar and carbohydrates, rather than from butter, meat, or other dietary fats.
2. Fructose is the strongest trigger for turning sugar into fat. Fructose (found in sugary drinks, fruit juice, and high-fructose corn syrup) bypasses normal control points in the liver and powerfully activates the process that converts carbohydrates into fat.
3. Insulin resistance keeps fat production switched “ON” in the liver. When your body becomes less sensitive to insulin, the liver continues making new fat even when it should be resting. This leads to higher blood fats and sugar levels at the same time — even while fasting.
4. Low-fat, high-carb diets can sometimes increase blood saturated fats. Cutting dietary fat but replacing it with refined carbohydrates and sugars can stimulate your liver to produce more saturated fat, which may explain why some people see their blood lipids worsen on classic low-fat diets.
5. Routine blood tests can reveal signs of increased fat production in the liver. Elevated fasting triglycerides (above 150 mg/dL), a high TG: HDL ratio (above 3.0), and other common lipid markers often indicate that your liver is actively converting excess carbohydrates into fat.
6. Carbohydrate restriction + fructose elimination is the most effective way to downregulate DNL. Reducing carbs (especially <130 g/day, or <50 g for ketosis), eliminating added sugars and liquid fructose, incorporating time-restricted eating, and exercising reliably suppresses DNL, improves MASLD, and lowers triglycerides.
The Saturated Fat Paradox Nobody Talks About
Imagine following a strict low-fat diet for six months — cutting butter, red meat, eggs, and cheese — only to have your doctor tell you that your blood saturated fat levels are still dangerously high.
This is not a hypothetical. It is a scenario played out in clinics around the world, and it has baffled both patients and physicians for decades.
The answer lies inside your liver, in a biochemical process called de novo lipogenesis (DNL) — literally, the creation of new fat from scratch. When you consume excess carbohydrates, especially refined sugars and fructose, your liver can do something remarkable and problematic: it converts those sugars into saturated fatty acids, primarily palmitic acid (C16:0), and releases them into your bloodstream.
The result is a paradox: your circulating palmitate levels reflect not how much butter you ate, but how much sugar your liver processed.
This article breaks down the full science of de novo lipogenesis — the molecular switches that turn it on, why fructose accelerates it, why it stays stuck in the "on" position in insulin-resistant individuals, and what you can do right now to dial it down. By the end, you will understand this process more deeply than most clinicians and have a clear, evidence-based roadmap for correcting it.
What you'll learn:
The step-by-step biochemistry of how sugar becomes saturated fat in your liver
The two master transcription factors that control this process
Why fructose is far more potent at driving DNL than glucose
How to interpret your bloodwork in light of DNL activity
Proven dietary and lifestyle interventions to suppress DNL and restore metabolic health
What Is De Novo Lipogenesis?
De novo lipogenesis (from the Latin: "making fat anew") is the metabolic pathway by which your body synthesizes fatty acids from non-fat precursors — chiefly carbohydrates. While the process occurs in several tissues, the liver (hepatic DNL) is by far the dominant site in humans, with adipose tissue playing a secondary role.
Under normal conditions, DNL is a low-level housekeeping process. Your liver uses it to store a small fraction of surplus carbohydrate energy as fat when glycogen stores are already full. In a healthy, active person eating balanced meals, this is not a problem.
The trouble begins when:
Carbohydrate intake is chronically excessive, especially from refined sugars and high-fructose corn syrup
Glycogen storage capacity is already saturated, leaving surplus glucose and fructose with nowhere to go but the DNL pathway
Insulin signaling is dysregulated, creating the biochemical environment that keeps the pathway permanently activated
According to a 2025 narrative review published in the journal Nutrients by Paoli et al., hepatic DNL is highly responsive to dietary carbohydrate intake and regulated by insulin via key transcription factors — and when dysregulated, contributes directly to the pathogenesis of metabolic dysfunction-associated steatotic liver disease (MASLD), the modern term for what was previously called NAFLD.
The primary product of DNL is palmitic acid (C16:0) — a 16-carbon saturated fatty acid. From palmitate, the liver can also produce:
Stearic acid (C18:0) via elongation
Palmitoleic acid (C16:1n-7) via desaturation by stearoyl-CoA desaturase 1 (SCD-1)
Oleic acid (C18:1n-9), another common DNL-derived fatty acid
These DNL-derived fatty acids are packaged into very-low-density lipoproteins (VLDL) and secreted into the bloodstream, where they contribute to hypertriglyceridemia, atherogenic dyslipidemia, and hepatic steatosis.
The Molecular Machinery: How Your Liver Converts Sugar to Fat
Understanding DNL at the biochemical level empowers you to make sense of why certain dietary and lifestyle choices have such a profound impact on metabolic health. Here is the enzymatic assembly line, step by step:
Step 1: Glucose Enters the Liver and Is Phosphorylated
After a carbohydrate-rich meal, glucose enters hepatocytes (liver cells) and is phosphorylated to glucose-6-phosphate (G6P) by the enzyme glucokinase. This traps glucose inside the cell and channels it toward glycolysis or glycogen synthesis.
Step 2: Glycolysis Produces Pyruvate
Glucose-6-phosphate proceeds through glycolysis, ultimately producing pyruvate. Pyruvate enters the mitochondria, where it is converted to acetyl-CoA by pyruvate dehydrogenase, and then condenses with oxaloacetate to form citrate via the TCA cycle.
Step 3: Citrate Is Exported to the Cytoplasm
When the cell has more acetyl-CoA than it can burn through oxidation, citrate is exported from the mitochondria into the cytoplasm. Here, the enzyme ATP-citrate lyase (ACLY) cleaves citrate back into oxaloacetate and cytosolic acetyl-CoA — the critical building block for fat synthesis.
Step 4: Acetyl-CoA → Malonyl-CoA (Rate-Limiting Step)
Acetyl-CoA carboxylase (ACC) converts acetyl-CoA to malonyl-CoA. This is the primary regulatory checkpoint of DNL — it is the rate-limiting step. Malonyl-CoA also serves a secondary role: it inhibits carnitine palmitoyltransferase 1α (CPT1α), the enzyme that shuttles fatty acids into mitochondria for oxidation. In other words, when your liver is making fat, it simultaneously blocks fat burning — a biochemical one-way valve.
Step 5: Malonyl-CoA → Palmitic Acid via Fatty Acid Synthase (FAS)
Fatty acid synthase (FASN) is a large, multifunctional enzyme complex that iteratively condenses acetyl-CoA and malonyl-CoA units. After seven cycles of condensation, reduction, dehydration, and further reduction, the result is a 16-carbon saturated fatty acid: palmitate (C16:0).
This palmitate is then esterified into triglycerides, packaged into VLDL particles, and exported into circulation — or accumulates in the liver itself, contributing to hepatic steatosis.
Key takeaway: Every excess gram of sugar that bypasses glycogen storage has the potential to become circulating palmitic acid through this six-step enzymatic cascade.
The Master Switches: ChREBP and SREBP-1c
Two transcription factors act as the master regulators of hepatic DNL. Understanding them is essential to understanding both the disease and the solutions.
ChREBP: The Carbohydrate Sensor
Carbohydrate Response Element-Binding Protein (ChREBP) is activated directly by intracellular glucose and fructose metabolites — no insulin signal required. When sugar floods the liver, glucose-6-phosphate and other glycolytic intermediates activate ChREBP, which then turns on the genes encoding the full DNL enzyme suite, including ACC and FASN.
Research published in PubMed has shown that ChREBP plays a central role in the paradox of selective hepatic insulin resistance: in obese, insulin-resistant states, the liver fails to suppress glucose production in response to insulin, yet ChREBP continues to drive lipogenesis — the worst of both metabolic worlds.
ChREBP exists in two isoforms:
ChREBPα: the full-length, glucose-sensitive form
ChREBPβ: a shorter, constitutively active form that lacks the glucose-inhibitory domain and keeps DNL running even at lower glucose concentrations
This is clinically important: in individuals with established metabolic dysfunction, ChREBPβ can maintain elevated DNL even between meals.
SREBP-1c: The Insulin-Responsive Switch
Sterol Regulatory Element-Binding Protein 1c (SREBP-1c) is the insulin-activated regulator of lipogenic gene expression. When you eat carbohydrates, insulin spikes and activates SREBP-1c via the AKT/mTORC1 signaling pathway. SREBP-1c then drives the transcription of ACC, FASN, and stearoyl-CoA desaturase (SCD-1), collectively ramping up the entire DNL machinery.
Crucially, in insulin-resistant individuals, hyperinsulinemia (chronically elevated insulin) provides a constant SREBP-1c stimulus — keeping DNL genes switched on 24 hours a day, seven days a week.
According to research in the Journal of Lipid Research (2025), both ChREBP (carbohydrate-driven) and SREBP-1c (insulin-driven) pathways work in concert to elevate DNL in overnutrition states — and each pathway independently supports increased fat synthesis even when the other is impaired.
Why Fructose Is the Turbocharger of DNL
Not all carbohydrates drive DNL equally. Fructose is uniquely and disproportionately potent at activating the DNL pathway — and this is a mechanistic fact, not a dietary opinion.
Here is why:
Fructose Bypasses the Primary Regulatory Checkpoint
In glycolysis, glucose is regulated at the phosphofructokinase (PFK) step — a tightly controlled valve that prevents the pathway from overflowing. Fructose bypasses PFK entirely.
In the liver, fructose is phosphorylated directly to fructose-1-phosphate by fructokinase, which then splits into glyceraldehyde and dihydroxyacetone phosphate — both of which feed directly into glycolysis below the PFK checkpoint. This means fructose enters the carbon pipeline unchecked, flooding the liver with acetyl-CoA precursors.
Fructose Activates Both Master Switches
Uniquely, fructose activates both ChREBP and SREBP-1c — directly and independently of insulin. Research shows that fructose administration rapidly increases hepatic hexose-phosphate levels, which activate ChREBP, while also stimulating SREBP-1c activity through insulin-independent mechanisms.
A 2025 study published in The Journal of Nutrition found that fructose in drinking water rapidly induced the upregulation of key lipogenic genes — including ACC, ACLY, ELOVL6, FASN, and SCD1 — confirming fructose's potent role as a lipogenic stimulus in the liver.
The Sucrose Problem
Table sugar (sucrose) is 50% glucose and 50% fructose. High-fructose corn syrup (HFCS) is 42–55% fructose. Research cited in the American Journal of Clinical Nutrition shows that isocaloric high-carbohydrate diets stimulate measurable increases in hepatic DNL — and that the fructose fraction is the primary driver.
Animal research published in PMC found that mice fed sucrose-palmitate diets accumulated significantly more hepatic lipid than those on starch-based diets, attributable to a "disproportionate rise in hepatic de novo lipogenesis" driven by sucrose.
Bottom line: When you drink a sugary soda, juice, or sweetened coffee, the fructose fraction is flowing directly into your liver's fat factory with virtually no regulatory braking system to slow it down.
Circulating Palmitate: Reading Your Blood Fats Correctly
Most people — and many clinicians — look at elevated circulating palmitic acid and assume it reflects dietary saturated fat intake. The science says otherwise.
DNL-Derived vs. Dietary Palmitate
Research from the Journal of Clinical Lipidology (2023) has made clear that circulating palmitic acid has two distinct sources:
Dietary palmitate: absorbed from foods like red meat, dairy, and palm oil via the gut, packaged into chylomicrons, and delivered to the bloodstream
DNL-derived palmitate: synthesized by the liver from carbohydrate substrates, packaged into VLDL, and released into circulation
The critical distinction is that DNL-derived palmitate responds to carbohydrate intake, not fat intake. Multiple studies have shown that low-fat, high-carbohydrate diets can significantly elevate circulating DNL-derived fatty acids, including palmitate.
Plasma DNL Biomarkers to Know
Research published in the Journal of the American Heart Association using the Cardiovascular Health Study cohort found that serial measurements of these blood fatty acids reflect DNL activity:
Palmitic acid (16:0) — Primary saturated fat produced by DNL; rises with excess sugar/fructose intake and insulin resistance.
Palmitoleic acid (16:1n-7) — Formed from palmitate via SCD-1; marker of active hepatic or adipose DNL.
Stearic acid (18:0) — Elongated product of palmitate; reflects ongoing DNL activity.
Oleic acid (18:1n-9) — Downstream monounsaturated fat derived partly from DNL and partly from dietary fat intake
Elevated plasma palmitate was associated with increased all-cause mortality, cardiovascular disease risk, and incident heart failure in this large prospective cohort study — making accurate interpretation of its source critically important for guiding clinical recommendations.
The 2023 Carbohydrate-Palmitate Connection in Humans
A randomized trial from Frontiers in Nutrition found that consuming 1 liter per day of sugar-sweetened soda over 24 weeks significantly increased DNL-derived fatty acids, including palmitate, in plasma phospholipids, cholesteryl esters, and triglycerides — with no change in dietary saturated fat intake.
This data powerfully demonstrates that circulating palmitate is a metabolic barometer of carbohydrate handling, not simply a measure of dietary fat consumption.
Insulin Resistance: The DNL Switch Stuck in the "On" Position {#insulin-resistance}
In metabolically healthy individuals, DNL operates in a pulsatile fashion — turning on postprandially and subsiding during fasting. Insulin resistance breaks this elegant off switch.
The Selective Insulin Resistance Paradox
Here is the cruel irony of metabolic disease: in insulin-resistant liver tissue, insulin's ability to suppress glucose production is lost, but its ability to activate SREBP-1c and drive lipogenesis is preserved.
This is called selective hepatic insulin resistance, and it creates a perfect storm:
Blood glucose stays elevated (insulin can't suppress liver glucose output)
But lipogenesis continues unchecked (insulin still activates SREBP-1c)
The resulting hyperinsulinemia further amplifies lipogenic gene expression
The consequence: an insulin-resistant liver is simultaneously overproducing glucose and overproducing fat — contributing to hyperglycemia, hypertriglyceridemia, hepatic steatosis, and cardiovascular risk all at once.
DNL Is Elevated Even in the Fasting State
In individuals with MASLD or significant insulin resistance, hepatic DNL is elevated not just postprandially but also during overnight fasting — a state in which DNL should be near zero in healthy individuals.
Studies using stable isotope tracers have shown that fractional DNL (the proportion of VLDL-triglyceride derived from DNL) can be three to five times higher in subjects with MASLD than in healthy controls — even after an overnight fast.
This finding has profound implications: it means that simply skipping breakfast does not reset the lipogenic machinery in a metabolically compromised liver. The switch is on 24/7 until the underlying insulin resistance is addressed.
De Novo Lipogenesis and Metabolic Disease
Dysregulated DNL is not an isolated phenomenon — it is a central driver in a cascade of metabolic disorders that collectively affect hundreds of millions of people worldwide.
MASLD / NAFLD
MASLD (Metabolic dysfunction-Associated Steatotic Liver Disease), formerly known as NAFLD, is now the most prevalent chronic liver disease globally, affecting an estimated 25–38% of the world population. Research consistently shows that DNL is significantly upregulated in MASLD, contributing to hepatic triglyceride accumulation.
A 2023 review in Liver Research noted that in-depth mechanistic analysis of DNL in non-alcoholic fatty liver disease has confirmed it as a primary driver of excess liver fat, alongside dietary fat delivery and adipose tissue lipolysis — with DNL's contribution rising from roughly 5% of liver fat in healthy individuals to up to 26% in subjects with NAFLD.
Research cited in PMC further demonstrated that sucrose-stimulated DNL is an important prerequisite for liver pathology, specifically implicating DNL-derived palmitate (C16:0) as a mediator of steatohepatitis (liver inflammation and injury) in experimental models.
Type 2 Diabetes
Chronically elevated DNL contributes to:
Hepatic steatosis → impaired insulin signaling in the liver
Increased VLDL secretion → hypertriglyceridemia and atherogenic dyslipidemia
Ceramide accumulation from excess palmitate → lipotoxicity and beta-cell dysfunction
Systemic insulin resistance propagated by excess circulating lipids
Cardiovascular Disease
The Journal of the American Heart Association study found that higher plasma levels of DNL-derived fatty acids were independently associated with increased cardiovascular mortality across 22 years of follow-up in older US adults — underscoring that this is not merely a laboratory curiosity but a clinically meaningful risk factor.
Why Cutting Dietary Fat Alone Won't Fix Elevated Palmitate
This is the crux of the clinical paradox — and one of the most important concepts in modern nutritional science.
If your elevated plasma palmitate is DNL-derived, reducing dietary saturated fat will have minimal impact on it.
Here is why this matters clinically:
A patient following a low-fat diet (15% of calories from fat) but consuming 55–65% of calories from carbohydrates — including refined grains, fruit juice, low-fat yogurt with added sugar, and sports drinks — may be running their hepatic DNL pathway at full throttle. Their plasma palmitate can remain elevated or even increase despite what looks like a "heart-healthy" dietary pattern on paper.
Research from the American Journal of Clinical Nutrition has confirmed that isoenergetic low-fat, high-carbohydrate diets stimulate measurable increases in de novo hepatic lipogenesis in both lean and overweight men, and that the primary DNL product, palmitate (C16:0), is elevated postprandially in a dose-dependent fashion to carbohydrate intake.
The Low-Fat Diet Trap
Here is the problem in practical terms:
You remove fat → you feel less satiated → you eat more carbohydrates
More carbohydrates → more substrate for DNL
More DNL → more hepatic palmitate synthesis
More hepatic palmitate → higher plasma triglycerides and palmitate
Clinician sees elevated triglycerides → recommends further fat restriction
This cycle has been playing out in clinical practice for 40+ years. Breaking it requires understanding DNL as the missing link between carbohydrate excess and elevated circulating saturated fat.
How to Downregulate DNL: Evidence-Based Strategies
The good news: DNL is a highly responsive, regulatable pathway. The following interventions have solid mechanistic and clinical evidence for suppressing hepatic DNL activity.
1. Carbohydrate Restriction
This is the most direct and potent intervention. By reducing the substrate supply (glucose, fructose) that feeds the DNL pathway, you simultaneously:
Reduce ChREBP activation (less intracellular glucose/fructose metabolites)
Reduce insulin secretion (less SREBP-1c activation)
Lower malonyl-CoA levels (reducing DNL flux)
Restore CPT1α activity (re-enabling fat oxidation)
A 2025 systematic review and meta-analysis published in PMC found that low-carbohydrate diets significantly reduced cardiovascular risk factors in patients with MASLD, in part by attenuating hepatic DNL via suppression of ChREBP. Studies typically define carbohydrate restriction as less than 130 g/day, with ketogenic diets (under 50 g/day) showing the most dramatic reductions in DNL activity.
Practical target: Reducing refined carbohydrate and added sugar intake is the first-line intervention. Even moderate carbohydrate reduction (from 55% to 35% of calories) produces measurable decreases in postprandial DNL.
2. Fructose Elimination
Given fructose's unique and potent ability to bypass regulatory checkpoints and activate both DNL master switches, targeted reduction of fructose is separately important from general carbohydrate restriction.
High-fructose foods to eliminate first:
Sugar-sweetened beverages (sodas, sports drinks, energy drinks, fruit juice)
High-fructose corn syrup (most processed snacks, condiments, and baked goods)
Agave nectar (up to 90% fructose)
Large amounts of concentrated dried fruit
Note: whole fruit in moderate quantities contains fiber that slows fructose absorption and does not appear to drive clinically significant DNL in most individuals.
3. Intermittent Fasting and Time-Restricted Eating (TRE)
A 2025 narrative review in Nutrients (Paoli et al.) specifically evaluated the effect of time-restricted eating on DNL and found that TRE offers a promising strategy to regulate hepatic DNL and improve metabolic health.
The mechanisms are multiple:
Reduced feeding window → fewer hours of postprandial insulin elevation → less SREBP-1c activation
Extended fasting periods → activation of AMPK → phosphorylation and inactivation of ACC (the rate-limiting DNL enzyme)
Glycogen depletion → carbohydrate consumed in the eating window preferentially fills glycogen stores rather than overflowing into DNL
Improved insulin sensitivity → less chronic hyperinsulinemia driving SREBP-1c
Common protocols with documented benefit include 16:8 (16 hours fasting, 8-hour eating window) and 5:2 approaches.
4. Aerobic and Resistance Exercise
Physical exercise has been shown to reduce hepatic DNL through multiple pathways:
AMPK activation → inhibits ACC → reduces malonyl-CoA → reduces palmitate synthesis
Improved insulin sensitivity → lowers basal insulin → reduces SREBP-1c-driven gene expression
Increased glycogen consumption → creates capacity for incoming carbohydrate → less substrate overflow into DNL
Enhanced mitochondrial β-oxidation → increased fat burning competes with fat synthesis
Even a single bout of moderate-intensity exercise significantly increases AMPK activity in the liver and skeletal muscle, creating a transient window of suppressed DNL.
5. Optimizing Hepatic Insulin Sensitivity
Since chronic hyperinsulinemia is a primary driver of SREBP-1c-mediated DNL, any intervention that improves insulin sensitivity will reduce basal DNL activity. Key strategies include:
Weight loss (even 5–7% body weight loss significantly improves hepatic insulin sensitivity)
Adequate sleep (sleep deprivation acutely worsens insulin sensitivity)
Stress management (cortisol promotes insulin resistance and can upregulate lipogenesis)
Omega-3 fatty acid supplementation (EPA/DHA can suppress SREBP-1c and SCD-1 gene expression)
Berberine and metformin (activate AMPK, reducing ACC activity)
Always consult your physician before starting any supplement or medication protocol.
Practical 7-Day DNL Reset Protocol
This evidence-based protocol is designed to rapidly reduce hepatic DNL activity. It is suitable for most healthy adults but should be reviewed with your doctor if you have any medical conditions.
Daily Nutrition Framework
Breakfast (10 AM) — High-protein, low-carb meal: eggs, avocado, leafy greens; fits a 16:8 time-restricted eating pattern.
Lunch (1 PM) — Protein with non-starchy vegetables and healthy fats: grilled salmon, broccoli, olive oil dressing.
Dinner (6 PM) — Protein-focused meal with vegetables and a small portion of complex carbohydrates if needed: chicken thighs, asparagus, lentils.
Optional Snack — Protein + healthy fat only: walnuts or a hard-boiled egg.
Beverages — Water, black coffee, or unsweetened green tea; avoid sugar-containing drinks.
Eliminate (7 Days)
All sugar-sweetened beverages
Fruit juice (all types)
Refined grains (white bread, pasta, white rice)
Added sugar in any form
Alcohol (directly stimulates DNL)
Daily Non-Negotiables
Exercise: 30+ minutes of moderate-intensity cardio or strength training
Sleep: 7–9 hours (sleep restriction raises insulin resistance by up to 30%)
Eating window: 8 hours maximum (16:8 TRE)
Fiber: 25–35g daily from vegetables and legumes to slow glucose absorption
Reassessment
After 7 days, reintroduce complex carbohydrates gradually (oats, sweet potato, legumes) while maintaining the elimination of sugar, refined grains, and fruit juice long-term.
Evidence Summary Table
Paoli et al., Nutrients (2025) — Exercise, ketogenic diets, and time-restricted eating reduce hepatic DNL, showing lifestyle interventions can directly suppress liver fat production.
Pi et al., PMC (2025) — Low-carbohydrate diets improved cardiometabolic risk factors in MASLD partly through suppression of ChREBP-driven DNL.
Vatner et al., Journal of Lipid Research (2025) — Multiple fuels, including glucose, lactate, and alanine, can sustain DNL during overnutrition, explaining persistent lipogenesis in metabolic disease.
Bhattacharjee et al., Journal of Nutrition (2025) — Just 14 days of fructose exposure markedly increased liver lipogenic enzymes including ACC, FASN, and SCD1.
Santos & Penha-Silva, Nutrition (2025) — Combined glucose and fructose intake synergistically amplifies hepatic DNL more than either sugar alone.
Rosqvist et al., Frontiers in Nutrition (2022) — Daily sugar-sweetened soda intake increased circulating DNL-derived palmitate despite no increase in dietary saturated fat intake.
Djoussé et al., JAHA — Higher blood biomarkers of DNL were associated with increased cardiovascular and all-cause mortality over long-term follow-up.
Softic et al., Digestive Diseases and Sciences (2016) — Fructose bypasses phosphofructokinase (PFK) and strongly activates both ChREBP and SREBP-1c pathways.
Ismail et al., PMC — Palmitate generated through DNL was more hepatotoxic than dietary palmitate, suggesting the source of saturated fat matters metabolically.
Common Myths and Mistakes
Myth 1: "My cholesterol panel doesn't test for this."
Reality: Standard lipid panels do not directly measure DNL activity. However, elevated triglycerides (especially fasting TG > 150 mg/dL), high VLDL cholesterol, low HDL cholesterol, and an elevated TG:HDL ratio (greater than 3.0) are reliable proxy markers of elevated hepatic DNL in a clinical setting. Ask your doctor about advanced lipid testing including plasma fatty acid profiles.
Myth 2: "Fruit is natural, so it can't be driving my liver fat."
Reality: While whole fruit contains fiber that moderates fructose absorption, very large quantities of fruit (especially in juice form, where fiber is removed) can contribute meaningful amounts of fructose to the liver. For individuals with established insulin resistance or MASLD, moderating even whole-fruit intake — especially high-fructose varieties like grapes, mangoes, and watermelon — may be warranted.
Myth 3: "I'm lean, so I don't need to worry about DNL."
Reality: Lean individuals can develop "metabolically obese" phenotypes with elevated hepatic DNL, particularly if they consume high amounts of sugar-sweetened beverages or fructose-rich foods. The key driver is carbohydrate load and insulin dynamics, not body weight alone.
Myth 4: "If I eat more saturated fat, my palmitate will go up."
Reality: At isocaloric intakes, adding dietary palmitate does not necessarily raise plasma palmitate the way that carbohydrate-driven DNL does — particularly when total energy is controlled. Research suggests that DNL-derived palmitate is more harmful to the liver than dietary palmitate, suggesting that the metabolic context in which palmitate is generated matters enormously.
Myth 5: "A low-fat diet is always heart-healthy."
Reality: A low-fat diet that replaces fat with refined carbohydrates can actively worsen the cardiometabolic risk profile by chronically stimulating DNL, raising triglycerides, lowering HDL, and increasing circulating palmitate — the opposite of what the diet intends to achieve.
Mistake 6: Testing DNL biomarkers after a single meal
Reality: Plasma DNL markers fluctuate significantly with acute meals. Meaningful biomarker assessment requires fasting samples and ideally longitudinal measurements over time, as used in the Cardiovascular Health Study. A single postprandial triglyceride spike tells you little about chronic DNL status.
Frequently Asked Questions
Q: What is de novo lipogenesis in simple terms?
De novo lipogenesis is your liver's built-in process for converting excess carbohydrates — primarily glucose and fructose — into saturated fat, specifically palmitic acid. When you eat more carbohydrates than your body can immediately use or store as glycogen, the overflow is converted into fat through this pathway and released into your bloodstream.
Q: Is de novo lipogenesis always bad?
No. DNL is a normal physiological process essential to development, cell membrane synthesis, and energy storage. It becomes problematic when it is chronically overactivated by excessive carbohydrate and fructose intake, insulin resistance, or metabolic disease, at which point it contributes to hepatic steatosis, hypertriglyceridemia, and cardiovascular risk.
Q: How much carbohydrate triggers de novo lipogenesis?
DNL is activated postprandially after any carbohydrate-containing meal, but the clinically significant increase in flux occurs when total carbohydrate intake exceeds glycogen storage capacity (roughly 400–500 grams in a typical person). Chronically consuming more than 250–350 grams of carbohydrate per day — especially from refined sources — has been shown to elevate DNL-derived VLDL triglycerides. The threshold varies considerably based on body composition, insulin sensitivity, and activity level.
Q: Does alcohol drive de novo lipogenesis?
Yes — significantly. Alcohol is metabolized in the liver to acetaldehyde and then to acetate, which directly generates cytosolic acetyl-CoA, the substrate for DNL. Alcohol also inhibits fatty acid oxidation by altering the NAD⁺/NADH ratio, creating a metabolic environment that strongly favors fat synthesis over fat burning. Alcoholic fatty liver disease shares many pathological mechanisms with carbohydrate-driven MASLD.
Q: Can de novo lipogenesis cause weight gain even on a calorie-controlled diet?
The relationship between DNL and weight gain is complex. DNL does not create energy from nothing — it converts carbohydrate calories into fat calories, which can then be stored. However, DNL also creates the hormonal and metabolic environment (via elevated insulin, elevated VLDL triglycerides, and suppressed fat oxidation through malonyl-CoA) that promotes fat storage and inhibits fat burning. These downstream effects on energy partitioning likely contribute to body fat accumulation beyond simple caloric accounting.
Q: What blood tests can reveal elevated DNL activity?
The most practical clinical indicators include: fasting triglycerides (> 150 mg/dL suggests elevated DNL), TG:HDL-C ratio (> 3.0 is a strong metabolic risk indicator), fasting insulin (elevated fasting insulin drives SREBP-1c), and hepatic ultrasound (which can reveal hepatic steatosis). Research-grade measurement uses stable isotope tracers (e.g., ¹³C-acetate infusion and MIDA analysis), which are not routinely available in clinical practice but are used in metabolic research studies.
Q: Is de novo lipogenesis reversible?
Yes — it is highly reversible with appropriate dietary and lifestyle modification. Studies have shown that as little as 2 weeks of carbohydrate restriction can significantly reduce hepatic fat content and DNL activity. Even moderate weight loss (5–10% of body weight) produces dramatic improvements in hepatic DNL, VLDL triglyceride output, and liver fat content.
Q: Do omega-3 fatty acids reduce de novo lipogenesis?
Yes, EPA and DHA (marine omega-3 fatty acids) have been shown to suppress hepatic DNL through multiple mechanisms: downregulation of SREBP-1c transcription, inhibition of SCD-1 (which desaturates DNL-derived palmitate), and activation of AMPK. Clinical trials have consistently shown that omega-3 supplementation lowers fasting triglycerides (a downstream marker of DNL), with doses of 2–4 grams per day of EPA+DHA showing the most robust effects.
Q: How does MASLD (fatty liver disease) relate to de novo lipogenesis?
MASLD is in part a disease of excessive DNL. While adipose tissue lipolysis and dietary fat delivery also contribute to hepatic fat accumulation, research has established that DNL's contribution rises from approximately 5% of liver fat in healthy individuals to 20–26% in those with NAFLD/MASLD — a four-to-five-fold relative increase. Targeting DNL through dietary carbohydrate restriction is one of the most evidence-supported interventions for reducing liver fat in MASLD.
Q: Should I get a DNL-specific test?
Currently, direct measurement of DNL (using stable isotope tracers) is available only in research settings. In clinical practice, the combination of fasting triglycerides, fasting insulin, liver enzymes (ALT, AST), TG:HDL ratio, and hepatic imaging provides a reasonable surrogate picture of DNL activity. Emerging research is exploring plasma phospholipid fatty acid profiles (particularly the 16:1/16:0 ratio as a marker of SCD-1 activity) as accessible clinical biomarkers of DNL.
Q: What medications target de novo lipogenesis?
Several pharmacological agents are under investigation or in clinical use:
Aramchol (SCD-1 inhibitor): shown to reduce liver triglycerides and improve liver histology in MASLD patients
ACC inhibitors (e.g., NDI-010976/firsocostat): block the rate-limiting step of DNL; in trials for MASLD/MASH
Metformin/berberine: activate AMPK → phosphorylate and inactivate ACC → reduce DNL flux
GLP-1 receptor agonists (e.g., semaglutide): improve insulin sensitivity, reduce SREBP-1c-driven lipogenesis, and are approved for MASLD-associated metabolic disease
Always consult a qualified healthcare provider before starting or changing any medication.
Clinician’s Perspective: When “Healthy Eating” Still Drives Fatty Liver
In clinical practice, many patients with elevated triglycerides and fatty liver insist they “hardly eat saturated fat.” A common scenario involves someone avoiding butter, red meat, and full-fat dairy while consuming large amounts of hidden sugars from fruit juice, smoothies, flavoured yoghurt, granola, sports drinks, snack bars, and refined “low-fat” foods.
Consider a typical patient: a 45-year-old office worker with fatigue, abdominal weight gain, and mildly elevated liver enzymes. His labs show triglycerides of 260 mg/dL, low HDL cholesterol, elevated fasting insulin, and ultrasound-confirmed fatty liver — despite following what he believes is a heart-healthy low-fat diet.
The explanation is often de novo lipogenesis (DNL). Excess refined carbohydrates and fructose overload the liver, which converts surplus sugar into saturated fat, particularly palmitate. In many patients, elevated circulating saturated fat reflects hepatic sugar processing more than dietary fat intake itself.
Clinically, this shifts the therapeutic focus. Simply cutting dietary fat while continuing high sugar intake may fail to improve metabolic health. More effective strategies typically include reducing refined carbohydrates, eliminating sugar-sweetened beverages, improving insulin sensitivity, increasing physical activity, and restoring healthy sleep patterns.
Medical disclaimer: This article is for educational purposes only and is not intended as medical advice. Always consult a qualified healthcare provider before making significant changes to your diet, supplement routine, or lifestyle, particularly if you have a known metabolic or liver condition.
Related Articles
Read the Full Guide: How to Reverse Hepatic Insulin Resistance Naturally
Small Dense LDL vs. Large LDL: Why Particle Size Matters More Than Your Cholesterol Number
Rethinking Dietary Fats: What New Research Reveals About Plant vs. Animal Fats | DR T S DIDWAL
What’s New in the 2025 Blood Pressure Guidelines? A Complete Scientific Breakdown | DR T S DIDWAL
Low-Fat vs. Low-Carb: Which Diet is Best for Weight Loss? | DR T S DIDWAL
5 Steps to Reverse Metabolic Syndrome: Diet, Habit, & Lifestyle Plan | DR T S DIDWAL
References
Benhamed, F., Denom, J., Real, X., Feuillet, G., Solgadi, A., Charifou, C., & Postic, C. (2016). ChREBP regulates fructose-induced glucose production independently of insulin signaling. The Journal of Clinical Investigation, 126(11), 4036–4048. https://doi.org/10.1172/JCI84583
Bhattacharjee, P., Fadlaoui, A., Ryan, C. E., Thackray, I. J., Morris, P. B., & Sunny, N. E. (2025). Induction of fructose mediated de novo lipogenesis coexists with the upregulation of mitochondrial oxidative function in mice livers. The Journal of Nutrition, 155(6), 1768–1781. https://doi.org/10.1016/j.tjnut.2025.04.012
Buziau, A. M., van de Wier, B., Schrauwen-Hinderling, V. B., Kooi, M. E., Schrauwen, P., & Hesselink, M. K. (2024). Hepatic glucokinase regulatory protein and carbohydrate response element binding protein attenuation reduce de novo lipogenesis. Molecular Metabolism, 87, Article 101984. https://doi.org/10.1016/j.molmet.2024.101984
Cross, E., Dearlove, D. J., & Hodson, L. (2023). Nutritional regulation of hepatic de novo lipogenesis in humans. Current Opinion in Clinical Nutrition & Metabolic Care, 26(2), 65–71. https://doi.org/10.1097/MCO.0000000000000898
Djoussé, L., Biggs, M. L., Lemaitre, R. N., King, I. B., Song, X., de Oliveira Otto, M. C., Siscovick, D. S., & Mozaffarian, D. (2020). Serial plasma phospholipid fatty acids in the de novo lipogenesis pathway and total mortality, cause-specific mortality, and cardiovascular diseases in the Cardiovascular Health Study. Journal of the American Heart Association, 9(13), Article e012881. https://doi.org/10.1161/JAHA.119.012881
Djoussé, L., Biggs, M. L., Lemaitre, R. N., King, I. B., Song, X., Siscovick, D. S., & Mozaffarian, D. (2020). Serial biomarkers of de novo lipogenesis fatty acids and incident heart failure in older adults: The Cardiovascular Health Study. Journal of the American Heart Association, 9(15), Article e014119. https://doi.org/10.1161/JAHA.119.014119
Horton, J. D., Goldstein, J. L., & Brown, M. S. (2002). SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. The Journal of Clinical Investigation, 109(9), 1125–1131. https://doi.org/10.1172/JCI15593
Hudgins, L. C., Parker, T. S., Levine, D. M., & Hellerstein, M. K. (2001). Postprandial de novo lipogenesis and metabolic changes induced by a high-carbohydrate, low-fat meal in lean and overweight men. The American Journal of Clinical Nutrition, 74(5), 565–567. https://doi.org/10.1093/ajcn/74.5.565
Ismail, O. Z., Zhang, X., & Al-Anany, M. A. (2015). Differential hepatotoxicity of dietary and DNL-derived palmitate in the methionine-choline-deficient model of steatohepatitis. Journal of Lipid Research, 56(6), 1150–1161. https://doi.org/10.1194/jlr.M056523
Kahn, C. R., et al. [See Note below]. (2022). Paradoxical activation of transcription factor SREBP1c and de novo lipogenesis by hepatocyte-selective ATP-citrate lyase depletion in obese mice. Proceedings of the National Academy of Sciences, 119(38), Article e2211325119. https://doi.org/10.1073/pnas.2211325119
Note: For citations with more than 20 authors, APA 7th requires listing the first 19 authors, then an ellipsis (...), and then the final author. If completing this for publication, replace "et al." with the explicit author sequence.
Koo, H. Y., Wallig, M. A., Chung, B. H., Nara, T. Y., Cho, B. H., & Nakamura, M. T. (2016). Isocaloric manipulation of macronutrients within a high-carbohydrate/moderate-fat diet induces unique effects on hepatic lipogenesis, steatosis and liver injury. The Journal of Nutritional Biochemistry, 28, 60–70. https://doi.org/10.1016/j.jnutbio.2015.09.022
Kratz, M., Marcovina, S., Nelson, J. E., Yeh, M. M., Kowdley, K. V., Callahan, H. S., & Kahn, S. E. (2010). Circulating palmitoleic acid and risk of metabolic abnormalities and new-onset diabetes. The American Journal of Clinical Nutrition, 92(6), 1350–1358. https://doi.org/10.1093/ajcn/2010.30074
Lee, D. E., McKay, L. K., Bareja, A., & White, J. P. (2026). Glutamine-driven reductive TCA cycle metabolism supports aged muscle stem cell function via de novo lipogenesis. Nature Aging, 6, 1007–1020. https://doi.org/10.1038/s43587-026-00912-x
Lee, D. S., et al. (2023). Tcf7l2 in hepatocytes regulates de novo lipogenesis in diet-induced non-alcoholic fatty liver disease in mice. Diabetologia, 66(5), 931–954. https://doi.org/10.1007/s00125-023-05875-1
Liu, Y., et al. (2017). LXRα regulates hepatic ChREBPα activity and lipogenesis upon glucose, but not fructose feeding in mice. Journal of Lipid Research, 58(8), 1563–1573. https://doi.org/10.1194/jlr.M074815
Ma, L., Robinson, L. N., & Towle, H. C. (2018). Interplay between ChREBP and SREBP-1c coordinates postprandial glycolysis and lipogenesis in livers of mice. Journal of Lipid Research, 59(4), 675–685. https://doi.org/10.1194/jlr.M082164
Noto, H. F., et al. (2023). Diet-derived and diet-related endogenously produced palmitic acid: Effects on metabolic regulation and cardiovascular disease risk. Journal of Clinical Lipidology, 17(4), 432–445. https://doi.org/10.1016/j.jacl.2023.05.002
Paoli, A., Cenci, L., Pompei, P., Baldari, C., & Bianco, A. (2025). The influence of physical exercise, ketogenic diet, and time-restricted eating on de novo lipogenesis: A narrative review. Nutrients, 17(4), Article 663. https://doi.org/10.3390/nu17040663
Parks, E. J., et al. (2024). Some paradoxes and unresolved aspects of hepatic de novo lipogenesis. npj Metabolic Health and Disease, 1, Article 12. https://doi.org/10.1038/s44324-024-00012-3
Pi, Y., Zhang, X., Zhang, L., et al. (2025). Low-carbohydrate diets reduce cardiovascular risk factor levels in patients with metabolic dysfunction-associated steatotic liver disease: A systematic review and meta-analysis of randomized controlled trials. Frontiers in Nutrition, 12, Article 12417190. https://doi.org/10.3389/fnut.2025.12417190
Rosqvist, F., Culshaw, K., Cooper, S., & Fielding, B. A. (2022). Effect of consuming 1 L/day of sugar-sweetened soda on de novo lipogenesis-derived fatty acids in plasma lipids. Frontiers in Nutrition, 9, Article 936828. https://doi.org/10.3389/fnut.2022.936828
Rosqvist, F., Iggman, D., Kullberg, J., Cedernaes, J., Johansson, H.-E., Neuman, A., & Risérus, U. (2020). High carbohydrate intake and circulating saturated fatty acids as markers of de novo lipogenesis and stearoyl-CoA desaturase activity in a Swedish population. European Journal of Nutrition, 59(7), 3245–3254. https://doi.org/10.1007/s00394-019-02058-6
Santos, H. O., & Penha-Silva, N. (2025). Revisiting the concepts of de novo lipogenesis to understand the conversion of carbohydrates into fats. Nutrition, 130, Article 112617. https://doi.org/10.1016/j.nut.2024.112617
Schulman, G. I., et al. (2013). Transcriptional control of hepatic lipid metabolism by SREBP and ChREBP. Seminars in Liver Disease, 33(4), 301–311. https://doi.org/10.1055/s-0033-1358525
Softic, S., Cohen, D. E., & Kahn, C. R. (2016). Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Digestive Diseases and Sciences, 61(5), 1282–1293. https://doi.org/10.1007/s10620-016-4054-0
Stefan, N., Fritsche, A., Weikert, C., di Giuseppe, R., Joost, H. G., Häring, H. U., Schulze, M. B., & Boeing, H. (2019). Circulating palmitoleic acid is an independent determinant of insulin sensitivity, beta cell function and glucose tolerance in non-diabetic individuals. Diabetologia, 62(11), 2004–2014. https://doi.org/10.1007/s00125-019-4965-0
Sunny, N. E., et al. (2023). Impact of sustained de novo lipogenesis on mitochondrial dysfunction and hepatocellular redox during fatty liver disease. American Journal of Physiology-Endocrinology and Metabolism, 324(2), E123–E135. https://doi.org/10.1152/ajpendo.00210.2022
Vatner, D. F., et al. (2025). Elevation of hepatic de novo lipogenesis in mice with overnutrition is dependent on multiple substrates. Journal of Lipid Research, 66(7), Article 100838.https://doi.org/10.1016/j.jlr.2025.100838
Zhu, Z., Zhang, X., Pan, Q., Zhang, L., & Chai, J. (2023). In-depth analysis of de novo lipogenesis in non-alcoholic fatty liver disease: Mechanism and pharmacological interventions. Liver Research, 7(4), 285–295. https://doi.org/10.1016/j.livres.2023.11.002