Beyond Glucose: Lipotoxicity as the Central Mechanism of Metabolic Disease
Lipotoxicity is fat buildup in organs like your heart, pancreas & kidneys. Learn how toxic lipids drive diabetes, heart failure & CKD — plus ways to reduce it.
METABOLISMDIABETES
Dr. T.S. Didwal, M.D.(Internal Medicine)
5/28/202624 min read
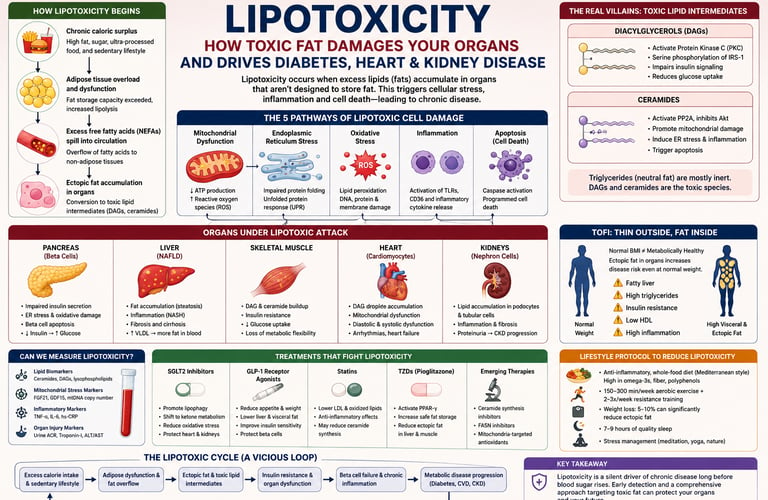
Lipotoxicity is the harmful buildup of fat in organs that are not designed to store it — such as the pancreas, liver, heart, and kidneys. When fat storage cells become overloaded, excess fatty acids spill over and turn into toxic molecules (like ceramides and diacylglycerols). These damage cells, trigger inflammation, and drive insulin resistance, type 2 diabetes, heart failure, and chronic kidney disease — often years before blood sugar becomes abnormal.
Key takeaways
1. Lipotoxicity is "fat in the wrong places"
When your body's fat storage system (adipose tissue) gets overwhelmed, excess fatty acids spill into organs not designed to hold fat—like the pancreas, liver, heart, and kidneys. This creates toxic lipid byproducts (especially DAGs and ceramides) that damage cells, trigger inflammation, and impair organ function—often years before blood sugar rises.
2. It drives type 2 diabetes through a vicious cycle
Toxic fat harms muscle (causing insulin resistance), overloads the liver, and slowly kills insulin-producing beta cells in the pancreas. This "gluco-lipotoxic" process explains why diabetes tends to progress over time. Beta cell loss is largely irreversible, which is why early intervention is critical.
3. It silently damages the heart and kidneys
In the heart, ectopic fat leads to stiffness (diastolic dysfunction) and eventually heart failure. In the kidneys, it injures filtering cells (podocytes) and promotes scarring, accelerating chronic kidney disease. These effects often occur independently of blood sugar levels.
4. You can have lipotoxicity even if you're lean (TOFI)
"Thin Outside, Fat Inside" means normal-weight people can still have dangerous fat accumulation in organs if their subcutaneous fat storage capacity is limited. Waist circumference, triglycerides, liver enzymes, and insulin levels are better warning signs than BMI alone.
5. SGLT2 inhibitors and GLP-1 agonists have powerful anti-lipotoxic effects
Beyond lowering glucose, these medications help clear toxic lipids from cells (via lipophagy), reduce inflammation, and shift metabolism to cleaner fuels. This explains their strong protective effect on heart and kidney health in people with diabetes.
6. Lifestyle changes are highly effective at reversing early lipotoxicity
Reducing saturated fat and refined carbs, increasing omega-3s and fiber, regular aerobic + resistance exercise, and even modest weight loss (5–10%) can dramatically lower ectopic fat, improve mitochondrial function, and restore metabolic health—especially when started early.
Introduction
Your fasting glucose is 94 mg/dL. Your HbA1c is 5.4%. Your doctor smiles and says, “perfect.”
But deep inside your pancreas, a single beta cell just failed.
Not from sugar. From fat.
This is lipotoxicity: the quiet accumulation of toxic lipids in organs that were never meant to store them. While your glucometer celebrates, diacylglycerols and ceramides are jamming insulin signaling in your muscle, scarring filtration units in your kidneys, and stiffening the walls of your heart.
For decades we blamed glucose. We measured it, managed it, and declared victory when it normalized. But a 2025 review in Cell Discovery argues we’ve been tracking the wrong enemy. Glucose is the smoke. Lipotoxicity is the fire. And the fire starts years before your blood sugar ever catches flame.
The most dangerous part? You can be lean. You can run marathons. You can have “great labs” and still be a TOFI — Thin Outside, Fat Inside. When your subcutaneous fat locker is full, excess lipids spill into your liver, heart, pancreas, and kidneys. Those organs don’t have storage units. They have mitochondria, endoplasmic reticulum, and DNA — all of which break under lipid overload.
Beta cell apoptosis is irreversible. Cardiac diastolic dysfunction rarely announces itself. Kidney fibrosis doesn’t hurt until it’s stage 3. By the time diabetes shows up on a lab report, lipotoxic damage has been compounding for years.
This article isn’t about sugar. You already know that story from our earlier pieces. This is about the fire your glucometer can’t see — and what 2024–2026 research says you can actually do about it.
1. What Is Lipotoxicity? The Science in Plain Language
Lipotoxicity refers to the cellular dysfunction and death caused by excessive accumulation of lipids (fats) in tissues that are not specialized for fat storage—such as the pancreas, heart, kidneys, liver, and skeletal muscle.
Your body has a beautifully designed fat management system. Adipose tissue (body fat) acts as a dedicated storage depot, safely packaging excess fatty acids as triglycerides. Under normal conditions, this system keeps circulating free fatty acids (NEFAs—non-esterified fatty acids) within a safe range, releasing them gradually as fuel.
The problem begins when this system is overwhelmed.
When caloric surplus is chronic, adipose tissue can reach its storage limit or become dysfunctional. Fatty acids then "spill over" into non-adipose organs. Unlike fat cells, these tissues have a limited capacity to oxidize or sequester lipids safely. The overflow gets converted into toxic bioactive molecules—and that's when cellular damage begins.
Think of it this way: Your adipose tissue is a storage locker. As long as the "junk" stays in the locker, your house stays clean. Lipotoxicity happens when the locker is full and fat starts piling up in your kitchen (liver) or your electrical panel (heart)—where it doesn't belong and actively breaks things.
Key Lipotoxic Mechanisms at a Glance
Mitochondrial dysfunction: Fatty acid overload saturates the mitochondria's ability to burn fat for energy, leading to impaired ATP production
Endoplasmic reticulum (ER) stress: Excess lipids disrupt protein folding, triggering cellular stress responses
Oxidative stress: Lipid peroxidation generates reactive oxygen species (ROS) that damage cell membranes, DNA, and proteins
Inflammation: Toxic lipid species activate innate immune receptors (TLRs, CD36), triggering inflammatory cytokine cascades
Apoptosis: Cells under severe lipotoxic stress undergo programmed cell death
2. From Lipotoxicity to Pan-Lipotoxicity: A Paradigm Shift
For years, lipotoxicity was treated as a single, uniform phenomenon—too much fat in a cell equals damage. But a landmark 2025 review published in Cell Discovery by Cheng and colleagues fundamentally reframed the field.
Their concept of pan-lipotoxicity recognizes that:
Different lipid species cause different types of damage. Saturated fatty acids (like palmitate), polyunsaturated fatty acids (PUFAs), cholesterol, lysophospholipids, and oxidized lipids each activate distinct molecular pathways. They are not interchangeable threats.
Tissue context determines the injury pattern. The same lipid molecule may cause apoptosis in a pancreatic beta cell, fibrosis in a kidney podocyte, and arrhythmia in a cardiomyocyte—each organ has its own lipotoxic fingerprint.
Multiple organ systems are affected simultaneously. Pan-lipotoxicity affects the nervous system (neuroinflammation and cognitive decline), vasculature (endothelial dysfunction and atherosclerosis), liver (steatosis and fibrosis), kidneys (glomerular damage), and heart (cardiomyopathy)—often in parallel.
Lipid Sensing: How Cells Detect the Threat
One of the most important insights from pan-lipotoxicity research is how cells "sense" dangerous lipids. Key receptors include:
Toll-like receptors (TLR4, TLR2): Normally, pathogens activate these; saturated fatty acids can mimic this signal, triggering sterile inflammation
CD36 (fatty acid translocase): Facilitates lipid uptake; overexpression accelerates lipid accumulation and oxidative stress
GPR120 and GPR40: G-protein coupled receptors that modulate lipid-related inflammatory signaling
This immune-like recognition of excess lipids explains why lipotoxicity has such a strong inflammatory component—your body is treating its own fat stores as a foreign threat.
3. The Liver–Muscle–Beta Cell Death Spiral
One of the most clinically important insights from recent lipotoxicity research is how organ dysfunction forms a self-amplifying feedback loop. Understanding this cycle explains why metabolic disease is so difficult to interrupt once it gains momentum.
Step 1: Hepatic Lipid Overproduction
When the liver is overwhelmed with fatty acid influx (from diet, adipose overflow, or de novo lipogenesis), it increases very low-density lipoprotein (VLDL) secretion, releasing more lipids into circulation. Non-alcoholic fatty liver disease (NAFLD/MASLD) is the visible result, but the systemic lipid burden is the real danger.
Step 2: Muscle Lipid Infiltration
Excess circulating fatty acids are taken up by skeletal muscle—the body's largest insulin-sensitive tissue. Here, they are converted into diacylglycerols (DAGs) and ceramides, bioactive molecules that directly sabotage insulin signaling (more on these below). The result: muscle becomes less responsive to insulin, a hallmark of type 2 diabetes.
Step 3: Beta Cell Failure
As peripheral insulin resistance develops, the pancreas compensates by producing more insulin. But the same elevated circulating fatty acids that impair muscle are also toxic to the beta cells trying to save the day. Chronic lipid exposure:
Impairs glucose-stimulated insulin secretion (GSIS)
Triggers ER stress and oxidative damage in beta cells
Ultimately causes beta cell apoptosis—irreversible loss of insulin-producing capacity
Step 4: The Vicious Cycle
Beta cell death → less insulin → higher blood glucose → more fat mobilization → more lipotoxic stress → more beta cell death.
According to Chen et al. (2025) in Diabetes, Metabolic Syndrome and Obesity, this self-perpetuating loop is why type 2 diabetes is better understood as a gluco-lipotoxic disease—the interplay of excess glucose and excess lipids creates a combined injury far greater than either alone.
Clinical Pearl: This is why treating only blood sugar is insufficient. You can normalize HbA1c and still have active lipotoxic damage silently destroying beta cells, kidneys, and cardiomyocytes.
4. Lipotoxicity and Type 2 Diabetes: Rewriting the Story
Type 2 diabetes has long been taught as a disease of glucose dysregulation. That framing is incomplete.
A comprehensive 2025 review by Chen and colleagues argues persuasively that lipotoxicity is not a secondary consequence of diabetes—it is a primary driver. Here's the key evidence:
Lipotoxicity Precedes Hyperglycemia
Intramyocellular and intrahepatic lipid accumulation—measurable by MRI spectroscopy—are elevated in individuals with insulin resistance before blood glucose rises to diabetic levels. By the time a fasting glucose of 126 mg/dL triggers a diabetes diagnosis, years of silent lipotoxic organ damage may have already occurred.
Lipotoxic Beta Cell Apoptosis Is Irreversible
Unlike insulin resistance (which is largely reversible), beta cell loss is permanent. Once a critical mass of beta cells is destroyed by lipotoxic stress, no amount of lifestyle intervention can restore them. This explains:
Why diabetes worsens progressively even with good glucose control
Why early intervention—before significant beta cell loss—is so critical
Why some people with impaired fasting glucose develop overt diabetes rapidly while others plateau
The "Double Hit" Hypothesis
Chronic elevated NEFAs deliver a double hit to the pancreas:
Functional impairment: Disrupted glucose sensing → reduced insulin secretion per beta cell
Structural destruction: ER stress and ROS-driven apoptosis → fewer total beta cells
Together, these two mechanisms explain the progressive nature of type 2 diabetes and the eventual need for insulin therapy in many patients.
5. How Toxic Fat Destroys Your Kidneys
Kidney disease affects over 850 million people worldwide, and its true cause is routinely misunderstood. While diabetes and hypertension are correctly identified as primary risk factors, a 2025 paper by Anumas and Inagi in Nephrology reveals that renal lipotoxicity is a major—and largely independent—driver of chronic kidney disease (CKD) progression.
Where Lipids Accumulate in the Kidney
The kidney is exquisitely vulnerable to lipotoxic injury because it is one of the most metabolically active organs in the body, demanding massive amounts of ATP to continuously filter blood.
Glomerular compartment:
Podocytes (the specialized filter cells) accumulate lipid droplets, causing effacement (flattening) of the foot processes that are essential for filtration
Glomerular endothelial cells suffer from lipid-induced oxidative stress and inflammation
Result: proteinuria (protein leaking into urine) and declining filtration capacity
Proximal tubules:
Lipid droplet accumulation impairs the energy-intensive process of nutrient reabsorption
Mitochondrial dysfunction reduces ATP availability
Lipotoxic inflammation promotes interstitial fibrosis
The fibrosis cascade: Tubular lipotoxicity triggers TGF-β signaling and myofibroblast activation, laying down scar tissue that permanently reduces kidney function—the defining feature of progressive CKD.
Analogy: Your kidneys are high-precision filters that need enormous energy to run. Fat accumulation in these filters is like pouring grease into a delicate engine—it clogs the system and destroys the power supply simultaneously.
Why This Matters Clinically
Current CKD management focuses heavily on blood pressure control and glucose reduction. These are necessary but insufficient. The Anumas & Inagi (2025) review argues that targeting renal lipid metabolism directly is the missing piece of kidney protection strategies.
6. Cardiac Lipotoxicity: The Hidden Driver of Diabetic Heart Failure
People with type 2 diabetes have a 2–4-fold higher risk of developing heart failure compared to the general population, even accounting for coronary artery disease and hypertension. For decades, this elevated risk was attributed primarily to atherosclerosis and hypertension. A 2025 study by Luong, Yang, and Kim in the Journal of Molecular and Cellular Cardiology reveals a different culprit: cardiac lipotoxicity.
How the Diabetic Heart Accumulates Fat
Under normal conditions, the heart primarily burns fatty acids (60–70%) and glucose for energy. In the diabetic state, several changes accelerate cardiac lipid accumulation:
Elevated circulating NEFAs (from insulin-resistant adipose tissue) flood the heart with more fatty acids than it can oxidize
Impaired insulin signaling reduces glucose uptake, forcing greater—and eventually excessive—dependence on fatty acid oxidation
Mitochondrial saturation leads to incomplete fatty acid oxidation and diversion of surplus lipids into triglyceride storage
Lipid droplet accumulation begins—initially adaptive, then destructive as toxic intermediates form
The Cascade of Cardiac Damage
Based on your layout, here is the progression of cardiac lipotoxicity broken down into clear, structured points matching each clinical stage:
Early Stage
Mechanisms: Lipid droplet accumulation and mitochondrial stress.
Clinical Manifestation: Subclinical diastolic dysfunction (early signs of stiffening in the heart muscle before obvious symptoms appear).
Intermediate Stage
Mechanisms: Reactive oxygen species (ROS) generation, ceramide accumulation, and altered calcium handling.
Clinical Manifestation: Symptomatic diastolic heart failure (the heart struggles to relax and fill properly, leading to noticeable symptoms like shortness of breath).
Advanced Stage
Mechanisms: Cardiomyocyte apoptosis (heart cell death), structural remodeling, and electrical instability.
Clinical Manifestation: Systolic dysfunction and arrhythmias (the heart loses its pumping power, alongside disruptions to its normal rhythm).Diastolic dysfunction (impaired relaxation) is typically the first sign. The heart becomes stiff and less able to fill properly between beats. This explains why many diabetic patients with heart failure have preserved ejection fraction (HFpEF)—a form of heart failure that is notoriously difficult to treat.
As lipotoxic damage progresses, systolic dysfunction develops: the heart loses contractile power and pumping efficiency falls.
SGLT2 Inhibitors as Cardiac "Housekeepers"
One of the most exciting discoveries in cardiology is that SGLT2 inhibitors (empagliflozin, dapagliflozin, canagliflozin) substantially reduce heart failure hospitalizations and cardiovascular death in diabetic patients—benefits far beyond their glucose-lowering effect.
The mechanism is becoming clearer: these drugs mitigate cardiac lipotoxicity by:
Promoting lipophagy (autophagic clearance of lipid droplets)
Shifting myocardial fuel preference toward ketone bodies (a more oxygen-efficient fuel)
Reducing oxidative stress and mitochondrial damage
Decreasing cardiac inflammation
This reframes SGLT2 inhibitors not merely as glucose-lowering drugs but as cellular housekeepers that clean toxic lipid accumulations from the heart.
7. The "TOFI" Phenomenon: You Can't Judge Metabolic Health by Weight Alone
One of the most practically important concepts in lipotoxicity research is TOFI: Thin Outside, Fat Inside.
BMI has been the dominant tool for assessing metabolic risk for decades. But BMI measures total body weight relative to height—it tells you nothing about where fat is located or whether that fat is metabolically active and toxic.
Why Normal-Weight Individuals Can Have Severe Lipotoxicity
Healthy subcutaneous fat is metabolically "safe"—it stores excess lipids away from vital organs. But not everyone has the same capacity for subcutaneous fat expansion. When subcutaneous storage is limited:
Even modest fat accumulation forces lipids into ectopic sites (liver, heart, pancreas, kidneys)
These organs lack the machinery to safely handle fat storage
Lipotoxic damage can be severe even at a normal BMI
Studies using MRI-based body composition analysis consistently find that visceral fat and ectopic organ fat are far better predictors of metabolic risk than BMI or total body weight.
Warning Signs of TOFI-Related Lipotoxicity
Even at a "healthy" weight, be alert to:
Elevated fasting triglycerides (>150 mg/dL)
Low HDL cholesterol (<40 mg/dL in men, <50 mg/dL in women)
Elevated liver enzymes (ALT, AST)
Increasing waist circumference
Insulin resistance on fasting insulin testing
Fatty liver on ultrasound
Key insight: The scale doesn't tell the whole story. A lean person with high visceral fat and poor metabolic flexibility may face greater lipotoxic organ risk than a heavier person with healthy subcutaneous fat distribution.
8. The Molecular Villains: DAGs, Ceramides, and How They Destroy Insulin Signaling
Understanding lipotoxicity requires going deeper than "fat in cells." The real culprits are specific bioactive lipid intermediates generated when fatty acid influx exceeds oxidative capacity.
Diacylglycerols (DAGs)
When excess fatty acids can't be fully oxidized or stored as triglycerides fast enough, they accumulate as diacylglycerols. DAGs are potent activators of protein kinase C (PKC) isoforms, particularly PKCθ and PKCε.
The molecular consequence:
PKC activation → serine phosphorylation of insulin receptor substrate-1 (IRS-1) → disruption of the PI3K–Akt signaling cascade → reduced GLUT4 translocation to the cell surface → impaired glucose uptake.
In simpler terms: DAGs teach cells to ignore insulin, not by damaging the insulin receptor itself, but by jamming the signal downstream of it.
Ceramides
Ceramides are sphingolipids generated when saturated fatty acids (especially palmitate) are combined with sphingosine. They exert lipotoxic effects through multiple mechanisms:
PP2A activation: Dephosphorylates and inactivates Akt, a central node of insulin signaling
Direct Akt inhibition: Ceramides can physically prevent Akt membrane association
Mitochondrial outer membrane permeabilization: Triggers cytochrome c release and apoptosis
Inflammatory amplification: Ceramide activates NF-κB, amplifying cytokine production
Why Triglycerides Are Not the Problem
A common misconception: elevated triglycerides in blood are a risk marker, not necessarily the toxic molecule. Intracellular triglycerides are largely inert storage molecules. The problem occurs when the synthesis of triglycerides cannot keep pace with fatty acid influx, allowing DAGs and ceramides to accumulate.
This is why some people with fatty liver (high triglycerides in hepatocytes) paradoxically have less liver damage than those with lower total hepatic triglycerides but higher DAG/ceramide ratios.
9. Biomarkers: Can We Measure Lipotoxicity in Patients?
A critical limitation of lipotoxicity research has been the diagnostic gap: we can measure blood lipids, but we cannot directly quantify lipotoxic damage in pancreatic beta cells, renal podocytes, or cardiomyocytes in living patients.
A 2024 bioinformatic study by Nie and colleagues in the Journal of Diabetes Research made significant progress by identifying gene and protein signatures specifically associated with lipotoxicity-related diabetic nephropathy.
Biomarker Categories Under Investigation
1. Specific lipid species:
Plasma ceramide profiles (elevated C16:0 ceramide correlates with insulin resistance)
Diacylglycerol species in skeletal muscle biopsies
Lysophosphatidylcholines as markers of oxidative lipid damage
2. Mitochondrial dysfunction markers:
Fibroblast growth factor 21 (FGF21): liver-secreted marker of mitochondrial stress
GDF15: stress-responsive cytokine elevated in metabolic disease
Mitochondrial DNA copy number in circulating cells
3. Inflammatory mediators:
Tumor necrosis factor-α (TNF-α)
Interleukin-6 (IL-6)
High-sensitivity CRP
4. Organ-specific injury markers:
Urine albumin-to-creatinine ratio (renal podocyte stress)
Cardiac troponin-I (cardiomyocyte damage)
Liver stiffness via elastography (hepatic fibrosis)
The Promise of Precision Medicine
When validated biomarker panels are available clinically, they could enable:
Early detection of lipotoxic organ damage before function declines
Risk stratification: identifying who needs more aggressive lipid-targeted therapy
Treatment monitoring: objectively measuring response to anti-lipotoxic interventions
Clinical trial enrichment: selecting patients most likely to benefit from experimental therapies
10. Treatments That Target Lipotoxicity
Current metabolic medications offer anti-lipotoxic benefits that extend well beyond their primary (glucose-lowering) mechanisms. Understanding these effects can help you and your doctor make more informed treatment decisions.
SGLT2 Inhibitors (Empagliflozin, Dapagliflozin, Canagliflozin)
Primary mechanism: Inhibit glucose reabsorption in the kidneys, lowering blood glucose
Anti-lipotoxic mechanisms:
Promote lipophagy (autophagic clearance of lipid droplets) in cardiomyocytes and renal tubular cells
Shift cardiac and renal metabolism toward ketone body utilization, reducing oxidative stress
Reduce renal lipid peroxidation and mitochondrial damage
Decrease ectopic fat in the liver, heart, and kidneys
Evidence: Major cardiovascular outcome trials (EMPA-REG OUTCOME, DAPA-HF, CREDENCE) demonstrate cardiac and renal protection that cannot be fully explained by glucose-lowering alone.
GLP-1 Receptor Agonists (Semaglutide, Liraglutide, Tirzepatide)
Primary mechanism: Stimulate insulin secretion; reduce appetite and body weight
Anti-lipotoxic mechanisms:
Improve insulin sensitivity, reducing fatty acid mobilization
Decrease hepatic VLDL output, lowering circulating NEFAs
Reduce hepatic steatosis and liver fat content
May directly protect beta cells from lipid-induced apoptosis
Evidence: The LEADER and SUSTAIN-6 trials showed significant cardiovascular risk reduction; imaging studies demonstrate substantial reductions in liver and visceral fat.
Statins
Primary mechanism: Inhibit HMG-CoA reductase, reducing cholesterol synthesis
Anti-lipotoxic mechanisms:
Reduce circulating LDL and oxidized lipoproteins that contribute to vascular lipotoxicity
Anti-inflammatory effects via reduction of prenylated signaling proteins
May reduce ceramide synthesis (an area of active research)
Thiazolidinediones (Pioglitazone)
Primary mechanism: PPAR-γ agonism, improving insulin sensitivity
Anti-lipotoxic mechanisms:
Promote expansion and differentiation of subcutaneous adipose tissue (the "safe storage" compartment)
Redirect lipid storage away from ectopic sites
Reduce hepatic and intramyocellular lipid accumulation
Caveat: Associated with weight gain, fluid retention, and bone fracture risk; use requires careful clinical judgment.
Emerging Therapies
Ceramide synthesis inhibitors: In preclinical development; directly target a key lipotoxic mediator
FASN inhibitors: Block de novo fatty acid synthesis
Mitochondria-targeted antioxidants (MitoQ, SS-31): Reduce ROS from overwhelmed mitochondria
Lipophagy inducers: Pharmacological promotion of autophagic lipid droplet clearance
11. Practical Lifestyle Protocol to Reduce Lipotoxicity
Pharmacological interventions are powerful, but lifestyle modification remains the foundation of lipotoxicity management.
Nutrition: The Anti-Lipotoxic Diet
Reduce:
Saturated fatty acids (especially palmitate from animal fat and tropical oils)—directly generate ceramides and DAGs
Refined carbohydrates—promote de novo lipogenesis in the liver
Fructose is particularly hepatotoxic, promoting hepatic steatosis and VLDL overproduction
Ultra-processed foods—contain combinations of saturated fat, sugar, and salt that maximise lipotoxic burden
Increase:
Omega-3 fatty acids (fatty fish, flaxseed, walnuts): EPA and DHA compete with saturated fatty acids, reduce TLR4 activation, and improve mitochondrial membrane composition
Dietary fiber (legumes, vegetables, whole grains): Improves insulin sensitivity; reduces post-meal fatty acid spikes via gut microbiome effects
Polyphenols (olive oil, berries, green tea): Activate AMPK and Nrf2, enhancing fatty acid oxidation and antioxidant defences
Mediterranean-style eating pattern: Consistently associated with reduced hepatic and cardiac fat accumulation in imaging studies
Physical Activity: Exercise as Lipotoxicity Medicine
Exercise is arguably the most potent anti-lipotoxic intervention available. Multiple mechanisms work simultaneously:
1. Increased Fatty Acid Oxidation
Mechanism: Upregulates CPT-1 (carnitine palmitoyltransferase-1) and PGC-1α (peroxisome proliferator-activated receptor-gamma coactivator-1alpha).
Lipotoxicity Benefit: Directly burns off accumulated intracellular lipid stores, preventing them from turning into toxic lipid intermediates.
2. Mitochondrial Biogenesis
Mechanism: Activates PGC-1α, which drives the creation of new, healthy mitochondria.
Lipotoxicity Benefit: Expands the cell's overall capacity to safely process and oxidize incoming fatty acids without causing mitochondrial overload.
3. Improved Insulin Sensitivity
Mechanism: Upregulates GLUT4 glucose transporters on the cell surface.
Lipotoxicity Benefit: Restores metabolic flexibility and reduces compensatory hyperinsulinemia (excess insulin), which otherwise promotes further fat storage.
4. AMPK Activation
Mechanism: Triggers AMPK (AMP-activated protein kinase), the body's primary cellular energy-sensing pathway.
Lipotoxicity Benefit: Shuts down de novo lipogenesis (the creation of new fat within the cell) and shifts the cell into an energy-burning state.
5. Enhanced Muscle Glucose Uptake
Mechanism: Stimulates glucose uptake via an insulin-independent contraction pathway during physical activity.
Lipotoxicity Benefit: Bypasses impaired insulin signaling pathways to clear glucose from the bloodstream, significantly lowering the overall glucolipotoxic burden on tissues.
Practical prescription:
Aerobic exercise: 150–300 minutes per week of moderate intensity (brisk walking, cycling, swimming). This is the primary driver of fatty acid oxidation.
Resistance training: 2–3 sessions per week. Increases skeletal muscle mass—the primary site of lipid disposal. Even a 10% increase in muscle mass meaningfully improves lipid clearance.
High-intensity interval training (HIIT): Particularly effective for reducing liver fat and improving mitochondrial density in short intervention periods (8–12 weeks).
Weight Management
Even modest weight loss of 5–10% produces dramatic reductions in:
Hepatic triglyceride content (often >30% reduction)
Pancreatic fat (may partially restore beta cell function)
Intramyocellular lipid accumulation
Circulating NEFAs and triglycerides
This is why weight loss remains one of the most powerful anti-lipotoxic interventions—it simultaneously reduces fatty acid influx and allows adipose tissue to regain its healthy storage function.
Sleep and Stress Management
Often overlooked but mechanistically important:
Poor sleep elevates cortisol and growth hormone, increasing fatty acid mobilization from adipose tissue
Chronic psychological stress activates the HPA axis, elevating cortisol → increased lipolysis → higher circulating NEFAs
Evidence: Short sleep duration (<6 hours) is independently associated with elevated intrahepatic lipid content and insulin resistance
Anti-Lipotoxic Lifestyle Checklist
Eat ≥5 servings of vegetables and fruit daily
Choose fatty fish 2–3× per week (or supplement omega-3s)
Limit saturated fat to <10% of total calories
Eliminate or minimize trans fats completely
Perform 150+ minutes of aerobic activity per week
Include resistance training 2–3× per week
Aim for 7–9 hours of quality sleep nightly
Practice stress reduction daily (meditation, yoga, nature exposure)
Maintain a healthy waist circumference (<94 cm for men, <80 cm for women)
Monitor triglycerides, HDL, liver enzymes, kidney function regularly
12. Evidence Summary: Key Studies at a Glance
Chen et al. – Diabetes, Metabolic Syndrome and Obesity (2025)
Focus: Lipotoxicity and Type 2 Diabetes Mellitus (T2DM).
Key Finding: Lipotoxicity is a primary driver—not just a side effect—of T2DM, with pancreatic beta-cell apoptosis (programmed cell death) serving as a central feature of the disease.
Cheng et al. – Cell Discovery (2025)
Focus: Pan-lipotoxicity.
Key Finding: Introduced the concept of pan-lipotoxicity, showing that different lipid species cause highly specialized, organ-specific cellular damage through unique, distinct mechanisms.
Anumas & Inagi – Nephrology (2025)
Focus: Renal (kidney) lipotoxicity.
Key Finding: Ectopic lipid deposition (fat accumulating where it shouldn't) in the kidneys drives Chronic Kidney Disease (CKD) progression. Current pharmacological therapies offer renoprotection largely by targeting these anti-lipotoxic pathways.
Nie et al. – Journal of Diabetes Research (2024)
Focus: Lipotoxicity biomarkers.
Key Finding: Pinpointed specific gene and protein biomarker signatures directly tied to lipotoxicity-related diabetic nephropathy (kidney disease).
Luong et al. – Journal of Molecular and Cellular Cardiology (2025)
Focus: Cardiac (heart) lipotoxicity.
Key Finding: Diabetic cardiomyopathy is heavily driven by fat buildup within the heart muscle (intramyocardial lipid accumulation). Notably, SGLT2 inhibitors provide a strong cardiovascular benefit by acting via anti-lipotoxic mechanisms.
Li et al. – Signal Transduction and Targeted Therapy (2026)
Focus: Comprehensive lipid metabolism overview.
Key Finding: A broad, high-impact review map linking lipid dysregulation to an array of systemic conditions, including cancer, cardiovascular disease (CVD), diabetes, neurodegeneration, and autoimmune diseases.
13. Common Myths and Mistakes About Lipotoxicity
Myth 1: "Lipotoxicity only affects obese people." False. Lean individuals can have severe lipotoxicity if their subcutaneous fat storage capacity is limited, their mitochondrial function is impaired, or they have a genetic predisposition. The TOFI phenomenon (thin outside, fat inside) is well-documented.
Myth 2: "If my LDL is normal, I don't have a lipid problem." LDL cholesterol measures a specific lipoprotein class—it says nothing about intracellular DAG and ceramide accumulation, the actual drivers of lipotoxic insulin resistance and organ damage. Standard lipid panels do not measure lipotoxicity.
Myth 3: "All dietary fat causes lipotoxicity." Not all fat is equal. Unsaturated fats (olive oil, avocados, nuts, fatty fish) are not strongly lipotoxic and may even be protective. Saturated fatty acids—especially palmitate—are the primary inducers of ceramide synthesis and TLR4 activation.
Myth 4: "Lipotoxicity is irreversible." While severe beta cell loss and advanced fibrosis may be permanent, significant reversal of ectopic fat accumulation, insulin resistance, and organ function is achievable with intensive lifestyle change and appropriate pharmacotherapy—particularly in earlier stages.
Myth 5: "My glucose is controlled, so my metabolic disease is managed." HbA1c normalization is important but insufficient. Active lipotoxic injury can continue even with excellent glucose control if ectopic fat accumulation is not addressed. This is why multi-factorial treatment approaches that include lipid-targeting therapies are increasingly recommended.
Myth 6: "Lipotoxicity is just NAFLD." NAFLD (now called MASLD) is one manifestation of hepatic lipotoxicity. But pan-lipotoxicity extends to the pancreas, kidneys, heart, skeletal muscle, and nervous system simultaneously. NAFLD is a visible signal; pan-lipotoxic organ damage may be far more widespread.
14. FAQs
Q1: What is the main difference between lipotoxicity and obesity?
Obesity is defined by excess total body fat; lipotoxicity is defined by the accumulation of fat in the wrong places—non-adipose organs not designed for fat storage. Obesity substantially increases lipotoxicity risk, but the two are not synonymous. You can have obesity with good metabolic health (metabolically healthy obesity), or lipotoxicity at a normal body weight (TOFI). The location and type of fat matters more than total fat mass.
Q2: Can lipotoxicity be tested for at my doctor's office?
Currently, no validated clinical test directly measures cellular lipotoxicity. However, several markers are useful proxies: liver enzyme levels (ALT, AST), hepatic fat on ultrasound or MRI, fasting triglycerides, fasting insulin (HOMA-IR), and urine albumin-to-creatinine ratio for renal involvement. Emerging ceramide plasma assays are available in some research settings and may enter clinical practice soon.
Q3: How long does it take for lipotoxicity to develop?
Lipotoxic damage accumulates gradually over years to decades, generally tracking with the duration and severity of caloric surplus, insulin resistance, and chronic low-grade inflammation. However, with severe dietary excess or extreme obesity, measurable ectopic fat accumulation can develop within weeks. Conversely, intensive lifestyle intervention can meaningfully reduce ectopic fat within 8–12 weeks.
Q4: Does intermittent fasting reduce lipotoxicity?
Evidence is encouraging. Intermittent fasting protocols (16:8, 5:2) promote autophagy including lipophagy (the cellular self-digestion of lipid droplets), reduce circulating NEFAs during fasting periods, and improve insulin sensitivity. Several small clinical trials show significant reductions in liver fat with time-restricted eating. However, long-term data are still limited, and fasting protocols are not appropriate for everyone (particularly those on insulin or with certain medical conditions).
Q5: Are omega-3 fatty acids protective against lipotoxicity?
Yes, through several mechanisms. EPA and DHA (marine omega-3s) compete with saturated fatty acids for incorporation into cell membranes and metabolic pathways, reducing ceramide synthesis. They also activate GPR120, an anti-inflammatory fatty acid receptor, and improve mitochondrial membrane fluidity, enhancing fatty acid oxidation efficiency. A 2025 review cited a consistent inverse relationship between marine omega-3 intake and markers of hepatic and cardiac lipotoxicity.
Q6: Why do SGLT2 inhibitors protect the heart and kidneys beyond glucose control?
SGLT2 inhibitors create a mild caloric deficit by causing glucosuria (glucose excretion in urine). They also shift the body toward ketone body production, which is a more oxygen-efficient cardiac fuel than fatty acids. Most importantly for lipotoxicity, they enhance autophagy and lipophagy in cardiomyocytes and renal tubular cells, clearing toxic lipid accumulations, and they reduce oxidative stress and inflammation in these organs. These pleiotropic effects explain their dramatic cardiovascular and renal benefits in outcome trials.
Q7: Is there a genetic component to lipotoxicity risk?
Yes. Genetic variation in adipose tissue expandability (how much fat you can safely store subcutaneously), mitochondrial fatty acid oxidation capacity, ceramide synthesis enzymes (like SMPD3 and CERT1), and lipid receptor expression all influence individual lipotoxicity risk. This helps explain why some people develop metabolic disease at relatively low BMI while others remain metabolically healthy at higher weights.
Q8: Can children and adolescents develop lipotoxicity?
Increasingly, yes. The epidemic of pediatric obesity, NAFLD, and insulin resistance in children means lipotoxic processes are beginning earlier in life than ever before. Imaging studies show hepatic steatosis in overweight children as young as 8–10 years. Early prevention—diet, physical activity, limiting ultra-processed food—is critical to prevent decades of lipotoxic organ damage from accumulating.
Q9: Does alcohol worsen lipotoxicity?
Significantly. Alcohol impairs mitochondrial fatty acid oxidation, promotes hepatic de novo lipogenesis, increases intestinal permeability (allowing lipopolysaccharide into the portal circulation, activating hepatic TLR4), and elevates circulating NEFAs. Heavy alcohol use dramatically accelerates hepatic lipotoxicity and steatohepatitis. Even moderate intake may worsen ectopic lipid accumulation in individuals with pre-existing insulin resistance.
Q10: What role does the gut microbiome play?
An emerging and fascinating area. Gut dysbiosis (disruption of healthy bacterial communities) is associated with increased intestinal permeability, greater absorption of lipopolysaccharides (which activate TLR4 and promote lipotoxic inflammation), altered bile acid metabolism (affecting lipid absorption and hepatic signaling), and changes in short-chain fatty acid production that influence hepatic lipogenesis. Microbiome-targeted interventions (prebiotics, probiotics, dietary fiber) are under active investigation as anti-lipotoxic strategies.
Q11: Can lipotoxicity cause neurological symptoms?
Yes. The concept of pan-lipotoxicity explicitly includes the nervous system. Lipid accumulation in cerebral vasculature promotes endothelial dysfunction; in neurons, ceramide accumulation induces apoptosis and neuroinflammation. Emerging research links metabolic syndrome, insulin resistance, and elevated saturated fat intake to increased dementia risk, reduced hippocampal volume, and impaired cognitive function—all consistent with lipotoxic mechanisms.
Q12: Should I get tested for lipotoxicity if I'm "healthy"?
If you have risk factors—family history of T2DM or cardiovascular disease, central obesity, elevated triglycerides, fatty liver on imaging, or a sedentary lifestyle—ask your doctor about expanded metabolic assessment beyond fasting glucose. Fasting insulin, HOMA-IR, liver enzymes, fasting triglycerides, and a waist circumference measurement are reasonable starting points that are widely available and underused.
15. Conclusion: Beyond Glucose—A New Paradigm for Metabolic Health
The science is clear: metabolic disease cannot be adequately understood—or treated—through a glucose-centric lens alone.
Lipotoxicity is not a side effect of diabetes. In many cases, it is a cause.
Long before blood sugar rises to diabetic levels, toxic lipid intermediates are silently infiltrating the pancreas, degrading beta cells, impairing insulin signaling in muscle, scarring kidney filters, and remodeling the heart. By the time a diagnosis is made, years of lipotoxic organ damage may have accumulated invisibly.
The paradigm shift demanded by this research is profound:
Diagnosis must extend beyond HbA1c and fasting glucose to include markers of lipotoxic organ stress
Treatment must target lipid metabolism, mitochondrial health, and inflammatory signaling—not just glycemia
Prevention must begin earlier, before ectopic fat accumulation reaches critical thresholds
The good news: lipotoxicity is modifiable. Dietary change, exercise, weight management, and appropriately selected medications can meaningfully reduce ectopic fat, restore mitochondrial function, and protect at-risk organs—especially when interventions begin early.
Your Action Steps
Know your lipotoxicity risk: Get fasting insulin, triglycerides, liver enzymes, urine albumin, and waist circumference measured—not just glucose
Upgrade your diet: Prioritize omega-3s, polyphenols, and fiber; minimize saturated fat and ultra-processed foods
Move your body daily: Both aerobic and resistance exercise are non-negotiable anti-lipotoxic medicines
Have a medication conversation: If you have T2DM, CKD, or heart disease, ask your doctor whether GLP-1 agonists or SGLT2 inhibitors are appropriate, given their anti-lipotoxic effects
Don't stop at glucose control: Achieving normal HbA1c is important, but insist on a comprehensive metabolic assessment that addresses lipid burden
Revisit your risk annually: Metabolic health can change. Regular monitoring allows early intervention before irreversible organ damage accumulates
Metabolic disease is not inevitable. But preventing it requires seeing clearly what is actually happening inside our cells—not just what is visible on a glucometer.
Disclaimer: This article is for educational and informational purposes only and should not be considered medical advice. Always consult with qualified healthcare professionals before making changes to your health regimen or treatment plan. The information presented reflects current research as of February 2026 and may be subject to change as new evidence emerges.
Related Articles
References
Anumas, S., & Inagi, R. (2025). Mitigating lipotoxicity: A potential mechanism to delay chronic kidney disease progression using current pharmacological therapies. Nephrology, 30(7), e70098. https://doi.org/10.1111/nep.70098
Chen, B., Li, T., Wu, Y., Song, L., Wang, Y., Bian, Y., Qiu, Y., & Yang, Z. (2025). Lipotoxicity: A new perspective in type 2 diabetes mellitus. Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy, 18, 1223–1237. https://doi.org/10.2147/DMSO.S511436
Chavez, J. A., & Summers, S. A. (2012). A ceramide-centric view of insulin resistance. Cell Metabolism, 15(5), 585–594. https://doi.org/10.1016/j.cmet.2012.04.002
Cheng, Y., Shao, S., Wang, Z., et al. (2025). From lipotoxicity to pan-lipotoxicity. Cell Discovery, 11, Article 27. https://doi.org/10.1038/s41421-025-00787-z
Note: For APA 7th edition, if a study has up to 20 authors, list all of them. If it has 21 or more, list the first 19, insert an ellipsis (...), and then list the final author. "Et al." is only used in text citations, not the reference list, unless the original metadata truly limits it.
Fazakerley, D. J., Krycer, J. R., Kearney, A. L., et al. (2019). Muscle and adipose tissue insulin resistance: Malady without mechanism? Journal of Lipid Research, 60(10), 1720–1732. https://doi.org/10.1194/jlr.R087510
Hotamisligil, G. S. (2017). Inflammation, metaflammation and immunometabolic disorders. Nature, 542(7640), 177–185. https://doi.org/10.1038/nature21363
Koves, T. R., Ussher, J. R., Noland, R. C., et al. (2008). Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metabolism, 7(1), 45–56. https://doi.org/10.1016/j.cmet.2007.10.013
Li, Z., Deng, W., Yang, L., et al. (2026). Lipid metabolism in homeostasis and disease. Signal Transduction and Targeted Therapy, 11, Article 55. https://doi.org/10.1038/s41392-025-02357-x
Luong, T. V. T., Yang, S., & Kim, J. (2025). Lipotoxicity as a therapeutic target in the type 2 diabetic heart. Journal of Molecular and Cellular Cardiology, 201, 105–121. https://doi.org/10.1016/j.yjmcc.2025.02.010
Marso, S. P., Daniels, G. H., Brown-Frandsen, K., et al. (2016). Liraglutide and cardiovascular outcomes in type 2 diabetes. New England Journal of Medicine, 375(4), 311–322. https://doi.org/10.1056/NEJMoa1603827
McMurray, J. J. V., Solomon, S. D., Inzucchi, S. E., et al. (2019). Dapagliflozin in patients with heart failure and reduced ejection fraction. New England Journal of Medicine, 381(21), 1995–2008. https://doi.org/10.1056/NEJMoa1911303
Muoio, D. M. (2014). Metabolic inflexibility: When mitochondrial indecision leads to metabolic gridlock. Cell, 159(6), 1253–1262. https://doi.org/10.1016/j.cell.2014.11.034
Nie, H., Yang, H., Cheng, L., & Yu, J. (2024). Identification of lipotoxicity-related biomarkers in diabetic nephropathy based on bioinformatic analysis. Journal of Diabetes Research, 2024, Article 5550812. https://doi.org/10.1155/2024/5550812
Ouwens, D. M., Diamant, M., Fodor, M., et al. (2005). Cardiac contractile dysfunction in insulin-resistant rats fed a high-fat diet is associated with elevated CD36-mediated fatty acid uptake and esterification. Diabetologia, 50(9), 1938–1948. https://doi.org/10.1007/s00125-007-0735-8
Samuel, V. T., Petersen, K. F., & Shulman, G. I. (2010). Lipid-induced insulin resistance: Unravelling the mechanism. The Lancet, 375(9733), 2267–2277. https://doi.org/10.1016/S0140-6736(10)60408-4
Summers, S. A. (2018). Could ceramides become the new cholesterol? Cell Metabolism, 27(2), 276–280. https://doi.org/10.1016/j.cmet.2017.12.003
Szendroedi, J., Phielix, E., & Roden, M. (2012). The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nature Reviews Endocrinology, 8(2), 92–103. https://doi.org/10.1038/nrendo.2011.138
Virtue, S., & Vidal-Puig, A. (2010). Adipose tissue expandability, lipotoxicity and the metabolic syndrome. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids, 1801(3), 338–349. https://doi.org/10.1016/j.bbalip.2009.12.006
Watt, M. J., & Hoy, A. J. (2012). Lipid metabolism in skeletal muscle: Generation of adaptive and maladaptive intracellular signals for cellular function. American Journal of Physiology-Endocrinology and Metabolism, 302(11), E1315–E1328. https://doi.org/10.1152/ajpendo.00547.2011
Zinman, B., Wanner, C., Lachin, J. M., et al. (2015). Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. New England Journal of Medicine, 373(22), 2117–2128. https://doi.org/10.1056/NEJMoa1504720
Last updated: February 2026. This article reflects current scientific evidence as of the publication date and is reviewed periodically. For personalized medical advice, consult a qualified healthcare professional.